Uworld 3
SGA
Infants are labeled small for gestational age (SGA) when their birth weight is below the 10th percentile for gestational age. Both maternal and fetal factors can lead to abnormal fetal growth. Maternal factors include preeclampsia, malnutrition, placental insufficiency, multiparity, or drug use. Fetal factors include genetic factors, chromosomal abnormalities, congenital infections, or inborn errors of metabolism. These low birth weight infants usually have intrauterine growth restriction, which can be symmetrical (height, weight, and head circumference are all equally affected) or asymmetrical (weight is affected more than height and head circumference).
Infants who are small for gestational age are at risk for several complications due to their size, including hypoxia, perinatal asphyxia, meconium aspiration, hypothermia, hypoglycemia, hypocalcemia, and polycythemia. The polycythemia results from increased erythropoietin secretion in response to fetal hypoxia.
Large for gestational age infants are at risk for developing hip subluxation and talipes calcaneovalgus primarily due to intrauterine deformation. Small for gestational age infants usually do not develop these abnormalities.
Hypoglycemia is commonly seen in small for gestational age infants due to decreased glycogen stores. Often, the hypoglycemia can be managed with early and frequent feeds. Infants with very low birth weights can have hyperglycemia due to low insulin secretion, but hypoglycemia would be more likely in an infant weighing 2.6 kg.
SGA infants have decreased subcutaneous fat and therefore impaired thermoregulation. As a result, these infants often become hypothermic.
Most SGA infants have hypocalcemia, which is thought to be caused by a decreased transfer of calcium across the placenta. Hypercalcemia, however, is not seen.
Congenital Toxoplasmosis
toxoplasmosis
Congenital toxoplasmosis
Risk factors
Raw or undercooked meatUnwashed fruits/vegetablesCat feces
Clinical features
MacrocephalyDiffuse intracranial calcificationsNonspecific signs of congenital infection (eg, jaundice, growth restriction, hepatosplenomegaly, blueberry muffin spots)
Diagnosis
Serology
Treatment
Pyrimethamine, sulfadiazine, folate
Congenital toxoplasmosis is caused by transplacental acquisition of Toxoplasma gondii from the mother. Maternal toxoplasmosis can be acquired by ingestion of cat feces, either directly (eg, exposure to kitty litter) or indirectly (via contaminated soil or produce). Toxoplasmosis can also be acquired by ingestion of raw or undercooked meat from animals infected with toxoplasmosis.
The majority of infants with congenital toxoplasmosis are asymptomatic at birth but experience chorioretinitis in adulthood due to reactivation of their infection. Infants who are symptomatic at birth may have macrocephaly, hydrocephalus, and diffuse intracerebral calcifications as seen in this infant. This infant's cholestasis and hepatomegaly are also consistent with a congenital infection but not specific for any particular pathogen.

Diagnosis is made by serology; the presence of infant IgM or IgA is confirmatory. Infected infants should receive pyrimethamine, sulfadiazine, and folate for a year; treatment reduces the parasite burden in the central nervous system and can relieve obstructive hydrocephalus.
Fetal Alcohol Syndrome
Fetal alcohol syndrome (FAS) is one of the leading preventable causes of birth defects and neurodevelopmental problems. Although in utero alcohol exposure may result in no apparent sequelae for some fetuses, others may suffer from FAS or be stillborn. Women who are pregnant or trying to conceive should be advised to abstain completely from alcohol as there is no known safe amount of prenatal alcohol consumption.
FAS is characterized by 3 pathognomonic facial dysmorphisms:
Small palpebral fissures
Smooth philtrum (vertical groove above the upper lip)
Thin vermilion border

Height and/or weight growth is compromised with percentiles <10th for age and sex. Microcephaly is often present, and these children suffer from cognitive and behavioral disorders. The phenotypic range of neurodevelopmental problems is wide and includes intellectual disability, attention-deficit hyperactivity disorder, social withdrawal, and delays in motor and language milestones. This child has appropriate gross motor skills for a 3-year-old, but his language is comparable to that of an 18-month-old. He also has socialization problems resembling autism spectrum disorder. Early diagnosis is critical for affected children to benefit from aggressive speech, physical, and occupational therapies.

Hypoplastic fingers/nails and cleft lip/palate are classic physical findings in fetal hydantoin syndrome. Pregnant women on phenytoin during their last trimester often receive prophylactic vitamin K to prevent neonatal bleeding as phenytoin may increase the rate of fetal vitamin K degradation. Phenytoin is not associated with the dysmorphic facies seen in this patient.
Septic Arthritis
septic arthritis

Precocious Puberty
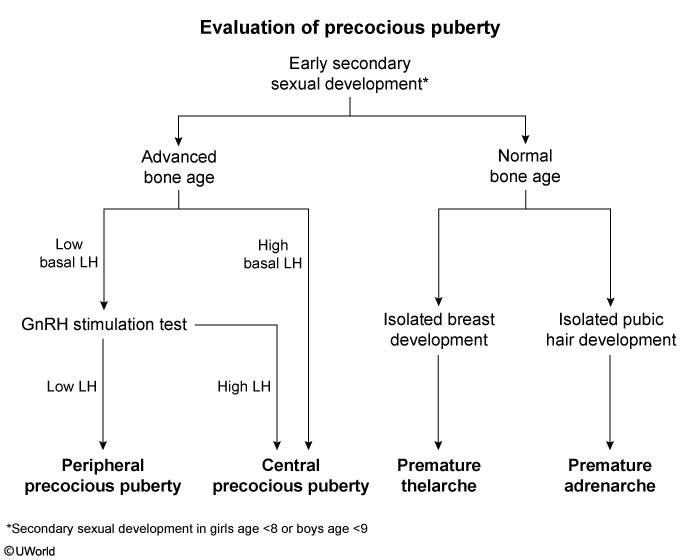
This patient has breast and pubic/axillary hair development consistent with precocious puberty, the onset of secondary sexual characteristics in girls age <8 and boys age <9. The initial evaluation of patients with precocious puberty is a bone age radiograph, which helps differentiate true precocious puberty from other causes of early pubic/axillary hair development (eg, premature adrenarche). Patients with true precocious puberty have increased estrogen/testosterone levels that accelerate skeletal maturation, resulting in advanced bone age and increased growth velocity (as seen in this patient).
The etiology of precocious puberty is categorized as either central (gonadotropin-dependent) or peripheral (gonadotropin-independent).
Central precocious puberty (CPP) results from early activation of the hypothalamic-pituitary-gonadal (HPG) axis. Pulsatile GnRH secretion stimulates elevated FSH and LH levels, as seen in this patient.
Peripheral precocious puberty (PPP) is caused by gonadal or adrenal release of excess sex hormones. Basal levels of FSH and LH are typically low due to negative feedback and remain low following GnRH agonist stimulation.
Patients with CPP require MRI of the brain to evaluate for a hypothalamic or pituitary tumor activating the HPG axis. If MRI is negative, the cause is most likely idiopathic precocious puberty, and GnRH therapy can be initiated. GnRH desensitizes the pituitary and suppresses FSH and LH secretion to slow pubertal progression and maximize height potential.
UTI

Urinary tract infections (UTIs) in infants and toddlers must be diagnosed and treated promptly as they usually involve the kidneys (pyelonephritis). Risk factors include girls at any age (short urethra), uncircumcised boys age <1, and underlying renal anomaly (eg, vesicoureteral reflux, posterior urethral valves). During infancy, symptoms are nonspecific and vague (eg, fever, fussiness, decreased urine output); abdomen/flank pain and dysuria can be difficult to recognize as infants are nonverbal. The presence of fever >39 C (102.2 F) in any child age <3 should prompt evaluation for occult UTI.
Serum blood urea nitrogen (BUN) and creatinine and urinalysis are quick, noninvasive, preliminary tests that should be done in all infants with illnesses involving the urinary tract. The BUN and creatinine provide a general sense of the patient's hydration status and degree of renal impairment. Urine dipsticks are also commonly performed, but they have a high rate of false-positive and negative results. Microscopic urinalysis is more accurate as it provides quantitative data on the degree of inflammation of the urinary tract (eg, number of white blood cells). A urine culture can identify bacteria type and antibiotic susceptibility. Patients who have received multiple antibiotic courses are at risk for resistant organisms.
A mid-stream clean-catch (Choice B) urine specimen is appropriate testing for patients who do not wear diapers. The external genitalia should be thoroughly cleaned to prevent contamination by skin flora. However, infants and toddlers in diapers should undergo straight catheterization of the urethra to obtain a sterile urine specimen. Clean-catch specimens are unreliable in diapered patients due to a high likelihood of stool or skin flora confounding the result.
Tinea Corporis
tinea coporis

Tinea corporis (ringworm) is a cutaneous dermatophyte infection most commonly caused by Trichophyton rubrum. Fungi thrive in humid climates and warm, moist areas (eg, shower surfaces, pools, gym mats, seats); therefore, sports involving skin-to-skin contact (eg, wrestling, gymnastics) are commonly implicated in the spread of infection.
Tinea lesions initially present as scaly, erythematous, pruritic patches that spread centrifugally. Untreated individuals, such as this patient, may develop a raised annular border and eventually central clearing as the fungus grows outward. Diagnosis is usually clinical, but skin scraping and potassium hydroxide examination are confirmatory for atypical or refractory cases.
First-line treatment for localized disease is a topical antifungal agent (eg, clotrimazole, terbinafine). Treatment should be continued until the lesion is resolved, which may take up to 3 weeks.
McCune Albright

This child has early breast and pubic hair development consistent with precocious puberty, the onset of secondary sexual characteristics in girls age <8 or boys age <9. Central precocious puberty is due to early activation of the hypothalamic-pituitary-gonadal axis. Peripheral precocity is attributed to premature secretion of sex hormones independent of GnRH.
In addition to precocious puberty, this patient has irregular café-au-lait macules confined to one side of the body and recurrent fractures due to polyostotic fibrous dysplasia, characteristic of McCune-Albright syndrome (MAS). MAS is a rare cause of precocious puberty due to a mutation in the GNAS gene, which results in constant G protein activation and overproduction of pituitary hormones. Therefore, in addition to GnRH-independent (ie, peripheral) precocious puberty (FSH, LH), MAS can also lead to thyrotoxicosis (TSH), acromegaly (GH), and Cushing syndrome (ACTH).
Seborrheic Dermatitis



This patient has features typical of seborrheic dermatitis (SD). Incidence of SD peaks in the first year of life and again in adulthood. SD is associated with colonization by Malassezia species and primarily affects areas with numerous sebaceous glands. In infants, these areas include the scalp ("cradle cap") (red circle), eyelids, nasolabial folds, postauricular area, and umbilicus.
The diagnosis of SD is based on characteristic examination findings of erythematous patches and plaques with yellow, oily scales, and mild pruritus may be present. Treatment is not always necessary as spontaneous resolution is common. First-line treatment for children includes gentle emollients and non-medicated shampoos. Widespread or recalcitrant SD is managed with low-potency glucocorticoid creams or topical ketoconazole.
Neonatal respiratory diseases

Persistent pulmonary hypertension of the newborn should be suspected in all term and post-term neonates with cyanosis. High pulmonary vascular resistance results in right-to-left shunting of deoxygenated blood through the foramen ovale and ductus arteriosus, resulting in hypoxia. The diagnosis is rare in very low birth weight infants. In addition, the x-ray findings in this patient are very typical of RDS.
Meconium aspiration syndrome occurs in term or post-term infants born through meconium-stained fluid. Meconium obstructs the airways and causes respiratory distress. X-ray would show patchy infiltrates, coarse streaking of both lung fields, and flattening of the diaphragm. This patient's history of prematurity and clear amniotic fluid makes this diagnosis unlikely.
Congenital Torticollis
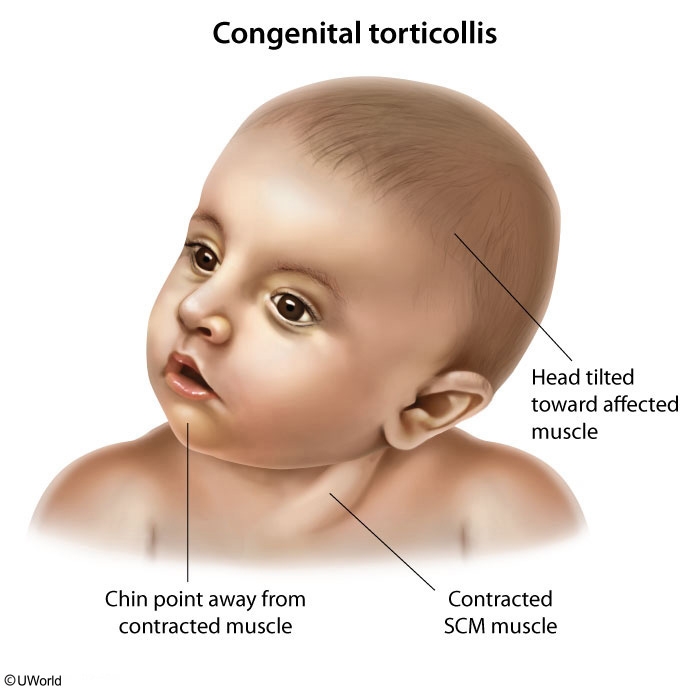
This infant presenting with a neck mass and head tilt to one side with chin deviation to the other has congenital muscular torticollis (CMT). CMT is a postural deformity that typically presents between age 1-6 months with limited range of motion of the neck. Physical examination may reveal a palpable, well-circumscribed mass that does not transilluminate in the inferior portion of sternocleidomastoid muscle.
Risk factors for CMT are related to crowding in the uterus, such as multiple gestation, breech positioning, and oligohydramnios. Associated conditions, which are also likely related to intrauterine positioning, include developmental dysplasia of the hip, metatarsus adductus, and clubfoot. Treatment strategies include positioning (eg, increased tummy time), passive stretching, and physical therapy. Missed or delayed diagnosis may lead to craniofacial asymmetry. Flattening of the head with ipsilateral anterior displacement of the ear and forehead, known as positional plagiocephaly, is also a common consequence of CMT.
RTA

This infant has evidence of a normal anion gap acidosis and failure to thrive. In the absence of an anion gap, a renal or gastrointestinal etiology of the acidosis is most likely. This child has no diarrhea but does have markedly alkalotic urine. These findings are suggestive of renal tubular acidosis (RTA). RTA is caused by a defect in the ability of the renal tubules to reabsorb bicarbonate (type 2 RTA) or excrete hydrogen (type 1 RTA). Type 1 RTA is often a genetic disorder and is commonly associated with nephrolithiasis. Type 2 RTA may be isolated but is more commonly a component of Fanconi syndrome (glucosuria, aminoaciduria, and phosphaturia are also present). Type 4 RTA is caused by a defect in the sodium/potassium exchange in the distal tubule, which results in hyperkalemic, hyperchloremic metabolic acidosis. In children, obstructive uropathy and aldosterone insufficiency are common causes.
All types of RTA can present as growth failure (due to poor cellular growth and division in acidic conditions). Screening laboratory results will show a low serum bicarbonate level and hyperchloremia, which lead to a normal anion gap metabolic acidosis. Evaluation of urine pH and urine electrolytes can help distinguish between the types of RTA. Given the markedly alkalotic urine in this patient, type 1 RTA is the most likely diagnosis. Treatment consists of oral sodium bicarbonate to normalize the serum bicarbonate levels.
Turner

This patient's short stature, signs of aortic coarctation (blood pressure differential), and absent menarche are most likely due to Turner syndrome (TS), a chromosomal abnormality caused by complete or partial loss of an X chromosome (45,X karyotype).
Patients with TS typically have ovarian dysgenesis, leading to "streak ovaries" (small ovaries with little to no follicles) and primary ovarian insufficiency. Because ovaries normally produce estrogen, patients with TS are estrogen deficient, which leads to amenorrhea and minimal or no breast development (thelarche). Estrogen also inhibits osteoclast-mediated bone resorption; therefore, patients with TS have increased bone resorption, decreased bone mineral density, and increased risk of osteoporotic fracture. Estrogen replacement therapy is given to girls with TS to promote normal sexual maturation and reduce the risk of osteoporotic fractures.
Migraine
Unlike migraine headaches in adults, migraines in children are often bifrontal and of shorter duration. Although migraines in children may be bifrontal, occipital headaches are extremely rare and, if present, should raise concern for a structural lesion.
First-line treatment of acute migraine headaches in children age <12 consists of supportive management (lying in a dark, quiet room with a cool cloth on the forehead) and administration of acetaminophen or a nonsteroidal anti-inflammatory agent such as ibuprofen. If the headache does not subside with these measures, oral, intranasal, or injectable triptans may be tried.
Indications for neuroimaging in a child with a headache include history of coordination difficulties; presence of numbness, tingling, or focal neurologic signs; history of a headache that causes awakening from sleep; or history of increasing headache frequency. This child has none of these.
Hypothyroidism
This clinical case is characteristic of congenital hypothyroidism, which may be familial or sporadic. The most common cause is thyroid dysgenesis (i.e. aplasia, hypoplasia, or ectopic gland), which has been incriminated in 85% of cases. Other causes include inborn errors of thyroxin synthesis (10%), and transplacental maternal thyrotropin-receptor blocking antibodies (5%). Infants initially appear normal at birth, but gradually develop apathy, weakness, hypotonia, large tongue, sluggish movement, abdominal bloating, and an umbilical hernia. Other signs include pathologic jaundice, difficult breathing, noisy respiration, hypothermia, and refractory macrocytic anemia. Infants initially appear normal due to the presence of moderate amounts of maternal hormones in the infant's circulation. For this reason, screening is mandated in all states at birth to allow for the early detection, treatment, and consequent improvement of the prognosis. Screening is done by measuring serum T4 and TSH levels. The treatment is levothyroxine (initial dose of 10 mcg/kg, then titrated accordingly).
Eczema Herpeticum
Eczema herpeticum is a complication caused by a superimposed primary herpes simplex virus (HSV) infection (usually type 1). An open area that is exposed to HSV type 1 can develop painful vesicles with an erythematous base that evolve to "punched-out" erosions with hemorrhagic crusting. The lesions can be localized or disseminated, and fever, irritability, and lymphadenopathy are typical. The infection may be life-threatening in infants; systemic acyclovir treatment should be initiated as soon as possible.
Physical examination shows an uncomfortable-appearing child with numerous painful, clear vesicles over erythematous skin on both cheeks as well as a few scattered lesions with overlying dark-red crusting:

Edward Syndrome
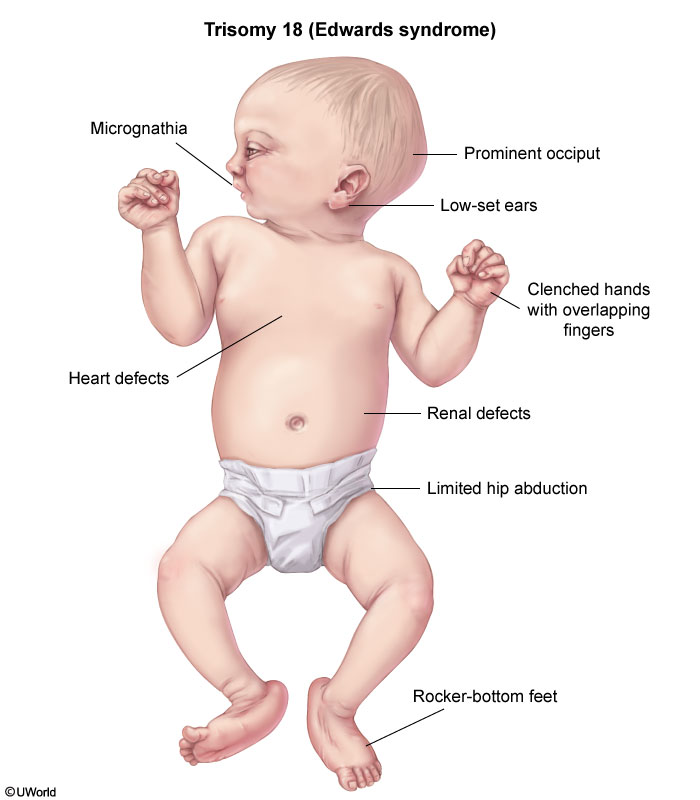
This patient's microcephaly, micrognathia, overlapping fingers, absent palmar creases, and rocker-bottom feet are consistent with trisomy 18 (Edwards syndrome). Congenital heart disease occurs in more than half of affected patients, with ventricular septal defect (VSD) being the most common abnormality. As pulmonary vascular resistance decreases after birth, VSD presents as a holosystolic murmur that is best heard at the left lower sternal border. Central cyanosis, which typically affects the trunk and mucous membranes, is not usually present with an isolated VSD, as evidenced by this infant's pink oral mucosa. This patient does have acrocyanosis, or peripheral cyanosis, which affects the hands and feet and is present in most healthy newborns.
The prognosis for trisomy 18 is very poor as most affected children die in the first month of life. Surgical repair of VSD improves survival, but those who survive are severely intellectually disabled.
Varicella Exposure

This child has been exposed to varicella-zoster virus (VZV) and should not be considered to have immunity (ie, 2 doses of vaccine or prior infection) since he did not receive his second VZV vaccine at age 4 years. Varicella is transmitted via airborne particles and is extremely infectious. Up to 90% of susceptible individuals will develop varicella after exposure to an infected person. Varicella is usually self-limited and mild, but serious complications include pneumonia, central nervous system disease (eg, cerebellar ataxia), and aggressive skin infections. These complications are more common in adolescents and adults, particularly those who are immunocompromised or pregnant.
Immunity to varicella is acquired by prior infection or by receiving 2 doses of VZV vaccine (at ages 1 and 4 years). Postexposure prophylaxis with VZV vaccine is indicated for this incompletely immunized child age >1 year who was exposed within the preceding 5 days. For susceptible individuals who cannot receive live-virus vaccines (eg, immunocompromised or pregnant patients), postexposure prophylaxis can be provided using varicella immunoglobulin. Infants (age <1 year) outside the neonatal period are not eligible for VZV vaccine and do not require immunoglobulin as they are at lower risk than neonates or older children.
Centor
FRONT

not good for preadolescent
preadolescent: must prove GAS before give abx
Acute Leukemia
Lymphoblasts lack peroxidase positive granules but often contain cytoplasmic aggregates of periodic acid Schiff (PAS) positive material. Immunostaining for terminal deoxynucleotidyltransferase (TdT) is positive in more than 95% of patients. TdT is expressed only by pre B and pre T lymphoblasts.
Myeloblasts on the other hand contain peroxidase positive material.
Eosinophilia causes
Neoplasm
Adrenal insufficiency
Allergy
Collagen vascular disease
Parasites
Growing Pains
Growing pains are a common musculoskeletal complaint in children, occurring in approximately 10%-30% of children age 2-12 years. The etiology of growing pains is unknown, but they are unrelated to growth, despite their name. The diagnosis of growing pains can be made clinically (Table) in the absence of systemic symptoms and abnormal examination findings. Laboratory studies and radiographs are not necessary.
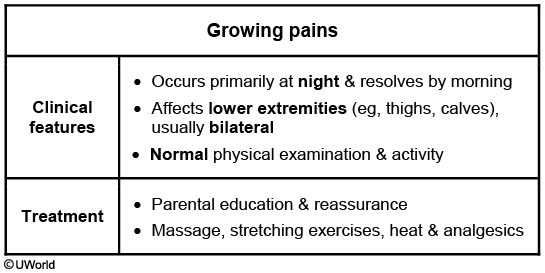
Treatment of growing pains consists of parental education and reassurance along with massage, muscle-stretching exercises, and administration of over-the-counter analgesics. Children with growing pains should be followed closely to monitor for pain that increases in frequency or intensity, which may warrant further evaluation.
SCFE
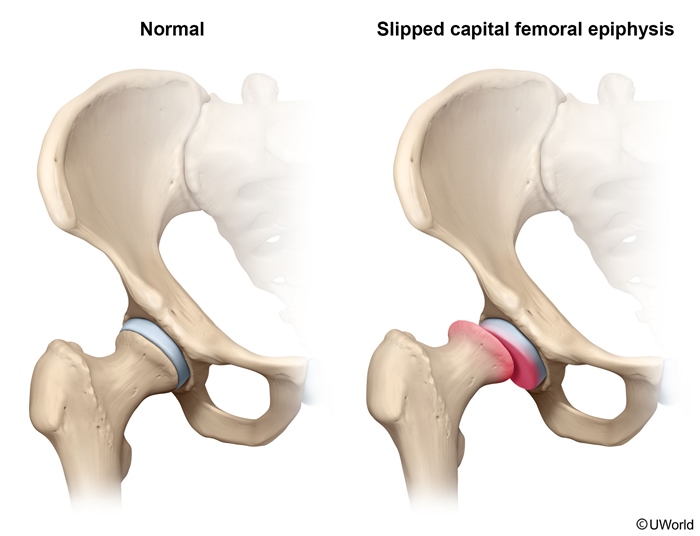
This patient has slipped capital femoral epiphysis (SCFE), displacement of the capital femoral epiphysis from the femoral neck. This typically presents in obese children age 10-16 years. Adolescent boys are affected slightly more often than girls. Additional risk factors include endocrinopathies (eg, hypothyroidism, growth hormone deficiency), renal failure, and radiation history. Children with endocrinopathies almost always have bilateral disease and present at an earlier age. Patients classically present with an insidious onset of dull hip or referred knee pain and altered gait with no preceding trauma. Minor trauma, as in this patient, can sometimes exacerbate the pain and bring the patient to medical attention. On examination, patients tend to hold the affected hip in passive external rotation and exhibit decreased internal rotation, abduction, and flexion. Diagnosis is made with plain radiographs of the hip (anteroposterior and frog-leg lateral views), which show the posteriorly and inferiorly displaced femoral head. Both hips should be imaged for comparison and to assess for contralateral displacement. The gold standard treatment is immediate surgical screw fixation at the current degree of slippage to avoid the risk of avascular necrosis (AVN).
Impetigo

The patient's rash is consistent with localized non-bullous impetigo. Impetigo is a common pediatric rash typically caused by Staphylococcus aureus or group A beta-hemolytic Streptococcus (Streptococcus pyogenes). Predisposing factors include a warm and humid climate, poverty/crowding, poor personal hygiene, and pre-existing skin trauma/inflammation (eg, insect bite, eczema). Colonization with staphylococci or streptococci is also a risk factor.
This superficial skin infection manifests with multiple painful pustules on the exposed areas of the face and extremities. Over the course of a week, the pustules rupture and harden into a characteristic golden-yellow ("honey") crust. Local lymphadenopathy can be present, but fever is unusual.
Antibiotics are indicated to reduce transmission and recovery time. Topical antibiotics (eg, mupirocin) are preferred for localized infection due to fewer side effects and less antibiotic resistance risk compared to oral therapy. Oral antibiotics (eg, cephalexin, dicloxacillin, clindamycin) (Choice B) are indicated when topical therapy is impractical for widespread non-bullous impetigo. Extensive bullous impetigo (ie, flaccid bullae containing yellow fluid) caused by S aureus is an additional indication for oral antibiotics. Thorough handwashing is also important to prevent the spread of this contagious infection.
Toxic
deferoximine: iron
calcium EDTA: severe lead
oral succimer: mild/moderate lead
zinc: copper
penicillamine: copper
Wilson Disease
Wilson disease
Pathogenesis
Autosomal recessive mutation of ATP7B → hepatic copper accumulation → leak from damaged hepatocytes → deposits in tissues (eg, basal ganglia, cornea)
Clinical findings
Hepatic (acute liver failure, chronic hepatitis, cirrhosis) Neurologic (parkinsonism, gait disturbance, dysarthria) Psychiatric (depression, personality changes, psychosis)
Diagnosis
↓ Ceruloplasmin & ↑ urinary copper excretion Kayser-Fleischer rings ↑ Copper content on liver biopsy
Treatment
Chelators (eg, D-penicillamine, trientine) Zinc (interferes with copper absorption)
The acute onset of psychotic symptoms (eg, bizarre behavior, auditory hallucinations) in a child or adolescent is rare. Medical and substance-induced causes must be ruled out, as they are potentially treatable. This patient's psychiatric symptoms (mood changes, irritability, bizarre behavior, auditory hallucinations), together with neurological symptoms (slurred speech, drooling, hand tremor) and abnormal liver function tests, are concerning for Wilson disease.
Wilson disease results in copper accumulation, most notably in the liver, brain, and cornea, and commonly manifests with hepatic involvement and/or neuropsychiatric symptoms in childhood or adolescence. Psychiatric symptoms may be subtle (eg, personality change, declining academic performance, odd behavior, irritability) or more severe (eg, depression, mania, psychosis). They can precede other manifestations and may be attributable to normal adolescentchanges or to primary psychiatric illness, leading to a delay in diagnosis and treatment. If Wilson disease is suspected, ceruloplasmin levels should be checked.
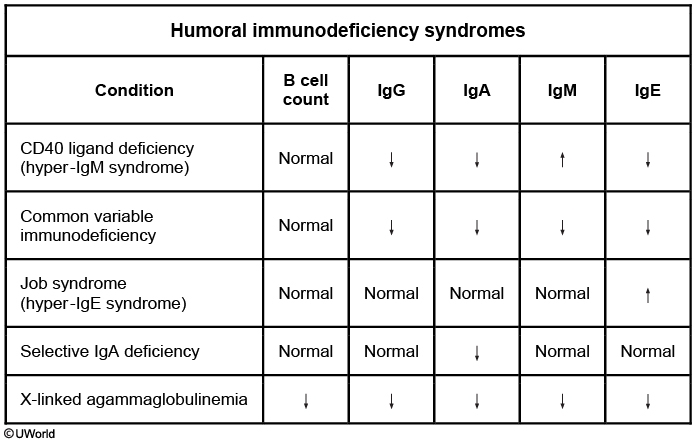
Ig levels
Friedreich Ataxia
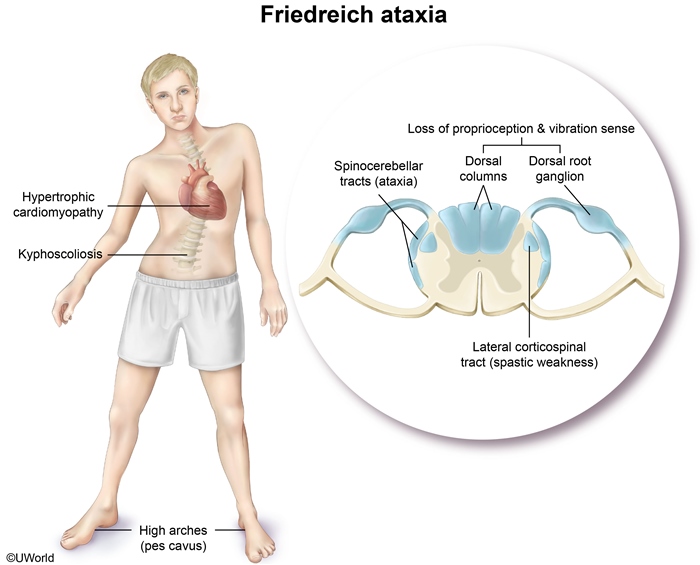
Friedreich ataxia (FA), the most common hereditary form of ataxia, is an autosomal recessive condition that classically presents with progressive ataxia in adolescence. FA is caused by an excessive number of trinucleotide repeat sequences (most commonly GAA) in the frataxin gene.
Neurologic findings include dysarthria, limb weakness, loss of deep tendon reflexes, and progressive gait and limb ataxia. Loss of position and vibratory sensesoccurs due to deterioration of the dorsal spinal column. Skeletal comorbidities include kyphoscoliosis and pes cavus (high-arched feet), as seen in this patient. Hypertrophic cardiomyopathy occurs in most patients, with an increased risk of arrhythmia and heart failure, usually leading to death by age 40. Imaging may reveal cervical spinal cord atrophy. Diagnosis is confirmed by genetic testing.
Management is supportive (eg, physical therapy, psychological support), as no disease-modifying therapies are available (Choice D). Prenatal genetic testing and counseling are available for future pregnancies for confirmed cases in siblings or if both parents are known carriers.
Friedreich ataxia
Osteosarcoma

The patient most likely has osteosarcoma, the most common primary bone tumor affecting children and young adults. Boys between ages 13 and 16 years are at higher risk. In children, the tumor occurs most frequently at the metaphyses of long bones such as the distal femur, proximal tibia, and proximal humerus. Constitutional symptoms such as fever, weight loss, and malaise are usually absent. On physical examination, the most important finding is a tender soft-tissue mass. Characteristic x-ray findings include a spiculated "sunburst" pattern (yellow arrow) and periosteal elevation known as the Codman triangle (red arrow). Alkaline phosphatase and lactate dehydrogenase are elevated from turnover of damaged osteocytes; high levels may correlate with adverse prognosis. Increased erythrocyte sedimentation rate is a non-specific marker of inflammation. Treatment includes tumor excision and chemotherapy.
Anemia of Prematurity

Anemia of prematurity (AOP) affects most preterm infants, and the onset and severity of anemia are proportional to the degree of prematurity. After delivery, circulating erythropoietin (EPO) normally decreases due to increased oxygen concentration in tissue. Decreased EPO causes decreased reticulocyte production in bone marrow. As a result, a physiologic red blood cell (RBC) nadir is expected and occurs at age 2-3 months in term infants. In preterm infants, however, low EPO levels are exacerbated by short RBC life span (40-50 days) and frequent phlebotomy in the neonatal intensive care unit. This can result in a significant, early-onset anemia.
Most infants with AOP are asymptomatic. Those who do have symptoms generally have mild tachycardia, increased apnea, or poor weight gain. AOP often is a diagnosis of exclusion; hemolysis, enzyme defects, hemoglobinopathies, and infection should be ruled out. Laboratory studies show decreased hemoglobin and hematocrit and a low reticulocyte count relative to the degree of anemia. The RBCs appear normal under light microscopy.
Treatment includes minimizing blood draws and ensuring adequate iron intake. RBC transfusions can be given if the infant is symptomatic but will further suppress EPO levels and delay recovery. Supplemental EPO is not effective in preventing the need for transfusions.
Sickle Cell
Despite vaccination, *S pneumoniae* remains by far the most common cause of sepsis in patients with SCD, usually from non-vaccine serotypes. Therefore, patients with SCD should receive prophylactic penicillin until at least age 5. Unfortunately, this patient was not taking penicillin.
Salmonella species and Staphylococcus aureus are the 2 most common causes of osteomyelitis in patients with SCD.
CHARGE
CHARGE syndrome: Coloboma, Heart Defects, Atresia choanae, Retardation of growth/development, Genito-urinary anomalies, and Ear abnormalities/deafness).
Primary Amenorrhea

Primary amenorrhea is the absence of menarche at age ≥13 in girls with no secondary sexual characteristics (eg, breast development). In those with secondary sexual characteristics, primary amenorrhea is absence of menarche at age ≥15. Amenorrhea can result from either functional or anatomic problems of the hypothalamus, pituitary gland, ovaries, uterus, or vagina. Therefore, the first step in management is to evaluate the female reproductive tract with a pelvic ultrasound to exclude anatomic abnormalities of the ovaries, uterus, or vagina.
Celiac
celiac disease
Classic celiac disease presents with gastrointestinal symptoms of abdominal pain, nausea, vomiting, diarrhea, and/or weight loss. Adolescents and adults may have extraintestinal symptoms such as fatigue, iron deficiency anemia (microcytic anemia, low ferritin), and dermatitis herpetiformis, as in this patient. Iron deficiency anemia in celiac disease is attributed to poor iron absorption secondary to duodenal villous atrophy. Iron supplementation (Choice E) can be considered after an underlying cause is confirmed and would likely improve anemia in celiac disease after initiation of a gluten-free diet. Dermatitis herpetiformis, a pruritic papular or vesicular rash associated with celiac disease, is located on the knees, elbows, forearms, and buttocks.
DDH
Age <4 months: Hip ultrasound
Age >4 months: Hip radiograph
Infants with abnormal examination should undergo bilateral hip ultrasonography. After age 4 months, when the femoral head and acetabulum are ossified, x-rays are preferred to evaluate acetabular development and positioning (Choice F). Confirmed DDH is treated with a Pavlik harness, a splint that holds the hip in flexion and abduction while preventing extension and adduction.

Bilious Vomiting

Trendelenburg sign

The physical exam finding described is the Trendelenburg sign, a drooping of the contralateral pelvis that occurs when the patient stands on one foot. The associated Trendelenburg gait is waddling in quality, caused by the trunk's rocking to compensate for this pelvic drooping during the stance phase of gait.
Normally, the gluteus medius and gluteus minimus muscles, which are both innervated by the superior gluteal nerve, function to abduct the thigh at the hip when standing on one foot or during normal ambulation when the body's weight rests on only one foot. Weakness of these muscles, as can occur in neuromuscular disease, impingement of or trauma to the superior gluteal nerve, or inflammatory myopathies, results in a positive Trendelenburg sign and gait.
DTAP

Mumps
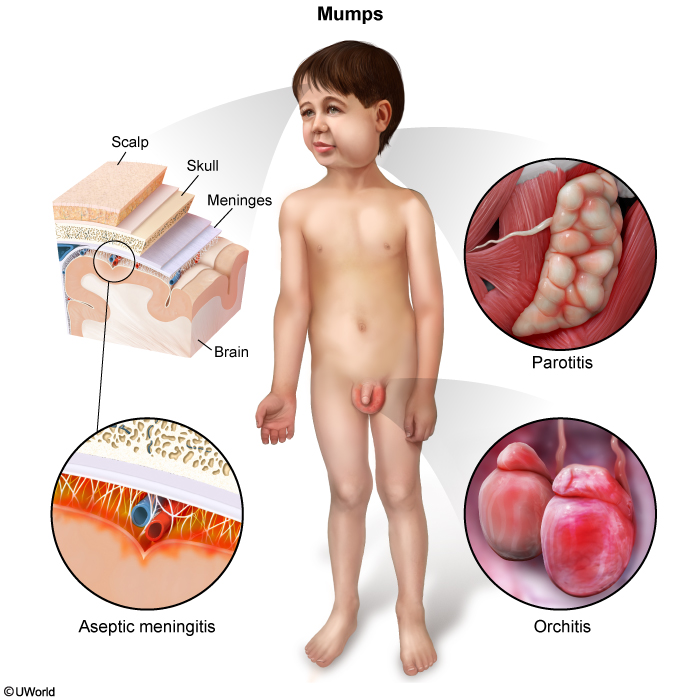
In general, mumps is a self-limited condition, but serious complications are possible. Aseptic meningitis is the most common complication of mumps, with cardinal symptoms including headache, fever, and nuchal rigidity. Orchitis is another potential complication that occurs primarily in postpubertal males and can impair fertility.
Perianal Dermatoses
This patient's presentation is most consistent with streptococcal perianal dermatitis, which is a superficial infection of Streptococcus pyogenes (group A Streptococcus) in the perianal region. This condition presents in infants and young children with bright, sharply demarcated, confluent erythema in the perianal or perineal region. Associated features include perianal pruritus and pain, particularly with stooling. In some patients, perirectal fissures and blood-streaked stools may occur. Due to pain while stooling, patients may also have constipation from withholding. Although patients often do not have concomitant streptococcal pharyngitis, there may be a close contact with recent streptococcal infection (eg, cellulitis, pharyngitis).
This condition can be suspected based on clinical features alone, and the diagnosis can be confirmed with a perianal bacterial culture. Treatment is with oral beta-lactam antibiotics (eg, penicillin, amoxicillin).
Perianal dermatoses
Diagnosis
Contact dermatitis
Candida dermatitis
Perianal Streptococcus
Epidemiology
Most common cause in infants
Second most common cause in infants
School-aged children
Examination
Spares creases/skinfolds
Beefy-red rash involving skinfolds with satellite lesions
Bright, sharply demarcated erythema over perianal/perineal area
Treatment
Topical barrier ointment or paste
Topical antifungal therapy
Oral antibiotics
Pediatric Stroke
Common etiologies of pediatric stroke
Sickle cell disease Prethrombotic disorders Congenital cardiac disease Bacterial meningitis Vasculitis Focal cerebral arteriopathy Head/neck trauma
This patient presents with acute, focal neurologic deficits, which should raise concern for stroke. Clinical features of stroke typically include focal weakness, hemiparesis, aphasia, seizures, or altered mental status. Although stroke is more common in older adults, it can occur in children, and the most common cause of pediatric stroke is sickle cell disease (SCD). In addition, this patient's history of adoption from Nigeria, a country with a high prevalence of SCD, raises the likelihood of SCD. SCD is an autosomal recessive disorder in which a mutation in the beta globin chain of hemoglobin results in hemoglobin polymerization, red blood cell deformation, and microvascular occlusion. Diagnosis of SCD is made via hemoglobin electrophoresis, which can confirm the presence of sickle hemoglobin.
Chronic vasoocclusion can result in endothelial damage, intimal proliferation, and eventual vascular stenosis, which increases the risk of cerebral ischemia, resulting in stroke. If concern for stroke exists, an MRI should be performed to confirm the diagnosis.
Vit A Def
This child's presentation is classic for Vitamin A deficiency. The condition usually manifests in the second or third year of life as impaired adaptation to darkness (which may progress to night blindness), photophobia, dry scaly skin, dry conjunctiva (xerosis conjunctiva), dry cornea (xerosis cornea) and a wrinkled, cloudy cornea (keratomalacia). Bitot spots (dry, silver-gray plaques on the bulbar conjunctiva) and follicular hyperkeratosis of the shoulders, buttocks, and extensor surfaces are less common findings.
Dry and Wet Beri Beri
Thiamine deficiency is associated with infantile and adult beriberi, as well as Wernicke-Korsakoff syndrome in alcoholics. Manifestations of infantile beriberi appear between the ages of two and three months and include a fulminant cardiac syndrome with cardiomegaly, tachycardia, cyanosis, dyspnea, and vomiting. Adult beriberi is categorized as dry or wet. Dry beriberi describes a symmetrical peripheral neuropathy accompanied by sensory and motor impairments, especially of the distal extremities. Wet beriberi includes this neuropathy in addition to cardiac involvement (eg, cardiomegaly, cardiomyopathy, congestive heart failure, peripheral edema, tachycardia).
G6PD
Glucose-6-phosphatase deficiency (type I glycogen storage disease, von Gierke disease) is caused by deficient glucose-6-phosphatase in the liver, kidneys, and intestinal mucosa. This results in impaired conversion of glycogen to glucose, leading to glycogen accumulation in the affected organs. Patients typically present at age 3-4 months with hypoglycemia (often resulting in seizures) and lactic acidosis (due to buildup in the liver). Other laboratory findings include hyperuricemia and hyperlipidemia. Physical examination demonstrates a doll-likeface with rounded cheeks, thin extremities, short stature, and a protuberant abdomen due to hepatomegaly. The spleen and heart are not involved.
PKU
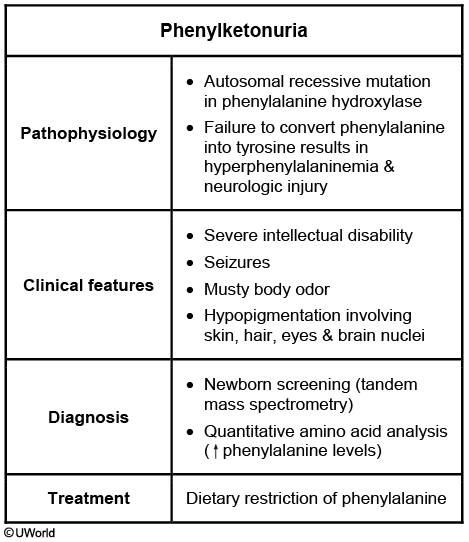
The clinical signs and family history of this patient are very suggestive of phenylketonuria (PKU). PKU is a rare (1/10,000 births) autosomal recessive disorder that is potentially devastating. A deficiency in the phenylalanine hydroxylase results in an inability to metabolize phenylalanine into tyrosine, which in turn causes an accumulation of phenylalanine and its neurotoxic byproducts. Untreated patients have severe intellectual disability and seizures. Other classic features include fair complexion and musty ("mousy") body odor, as in this patient.
Newborn screening is extremely important as early diagnosis and treatment can allow these patients to live relatively healthy lives with normal intellect. Most infants with PKU are asymptomatic initially; diagnosis is typically made from positive newborn screening tests. Tandem mass spectrometry of dried blood spots can detect the presence of metabolic products of phenylalanine. Mass spectrometry screening for PKU is mandated in all 50 states and is the most cost-effective screening test. If PKU is suspected later in life, as in this patient, quantitative amino acid analysis will show elevated phenylalanine levels.
Phenylalanine is an essential amino acid, and small amounts are necessary for growth and development. Treatment consists of a low-phenylalanine diet. Cereals, starches, fruits, vegetables, and phenylalanine-free milk formulas are recommended. High-protein foods should be avoided. Early diagnosis and treatment improve the prognosis, with most treated patients having normal mental development and a normal life span.
Dermatitis

Atopic dermatitis (eczema) is a very common condition characterized by pruritus, erythema, and scaly lesions on the skin. Pathogenesis involves epidermal dysfunction due to improper synthesis of stratum corneum components. Allergens can enter the disrupted skin barrier and generate an inflammatory response. Excessive bathing, dry environments, stress, overheating, and irritating detergents can trigger flares.
Eczematous lesions usually begin with pruritus alone and evolve to erythematous papules and scaly plaques. Severe lesions may have serous exudates and crusting. Infants typically have lesions in the distribution of the face, scalp, and extensor surfaces of the extremities. The lesions can also be seen in flexural creases in older children and adults.
Treatment includes trigger avoidance, frequent application of thick bland emollients, and use of hypoallergenic cleansers for bathing and laundry. Moderate and severe eczema may require topical anti-inflammatory ointments (eg, hydrocortisone).
(Choice A) Contact dermatitis is also characterized by an itchy red rash. In contrast to atopic dermatitis, lesions have indistinct margins and occur only in areas of direct allergen contact. It is common in older children and adults after sensitization to an allergen (eg, poison ivy resin, nickel, neomycin/bacitracin).
OI
Osteogenesis imperfecta
Pathogenesis
>90% autosomal dominant Type I collagen gene (COL1A1) defect
Clinical features
Mild to moderate Frequent fractures Blue sclera Conductive hearing loss Short to normal stature Dentinogenesis imperfecta Joint hypermobilityLethal (type II) In utero and/or neonatal fractures Pulmonary failure
This patient has osteogenesis imperfecta (OI), an autosomal dominant connective tissue disorder commonly caused by a type I collagen mutation. Type I collagen is a critical structural component of tissues in the bone, sclerae, skin, and teeth. OI has a varying spectrum of severity, from mild (type I) to lethal (type II) disease.
Due to defective collagen in osteoid (extracellular bone matrix), patients often begin having frequent fractures from minor trauma once they become mobile (around age 1). Many patients with OI also have dentinogenesis imperfecta, an opalescent blue-gray or yellow-brown discoloration of the teeth caused by discolored dentin shining through translucent and weak enamel. Joints are often hypermobile due to ligamentous laxity. Scleral thinning can lead to the appearance of blue sclerae; other manifestations depend on the severity of the disorder and may include hearing loss and short stature.


Osgood Schlatter
Osgood-Schlatter disease is a common cause of knee pain, particularly in adolescent male athletes. During early adolescence (typically ages 13-14 for affected males, and ages 10-11 for affected females), there are periods of rapid growth in which the quadriceps tendon puts traction on the apophysis of the tibial tubercle where the patellar tendon inserts. This traction apophysitis is worsened by sports that involve repetitive running, jumping, or kneeling, and it improves with rest. Approximately one fourth of affected individuals have bilateral disease. On physical examination, there is edema and tenderness over the tibial tubercle. A firm mass can sometimes be felt due to heterotopic bone formation. Pain can be reproduced by extending the knee against resistance. Radiographic findings are nonspecific and include anterior soft tissue swelling, lifting of tubercle from the shaft, and irregularity or fragmentation of the tubercle. Treatment consists of activity restriction, stretching exercises, and non-steroidal anti-inflammatory medications.

Prepatellar bursitis occurs with chronic irritation of the anterior knee. Symptoms include pain with direct pressure and superficial swelling over the patella.
(Choice C) Patellar tendonitis is an overuse syndrome resulting from repetitive jumping or kicking. Patients present with anterior knee pain after exercise. Unlike Osgood-Schlatter disease, patients have point tenderness at the inferior pole of the patella.
(Choice D) Tibial osteomyelitis is a bone infection, usually bacterial in origin. Symptoms include pain, swelling, tenderness, and erythema. Patients classically present with refusal to bear weight on the affected extremity. Systemic symptoms may also be present. The pain from osteomyelitis does not remit with rest.
(Choice E) Patellofemoral stress syndrome is an overuse injury commonly seen in runners. Patients present with anterior knee pain that worsens upon descending steps or hills. Pain is localized to the patella and radiographs do not demonstrate separation at the tibial tubercle.
Patellofemoral pain syndrome causes chronic anterior knee pain. Patients present with pain worsened by activity or prolonged sitting (due to sustained flexion) and may also have crepitus with motion of the patella.
Pes anserinus syndrome (often called anserine bursitis) is characterized by pain and tenderness at the anterior medial knee distal to the joint line. Iliotibial band syndrome causes lateral pain with tenderness at the lateral femoral condyle. These chronic overuse injuries do not cause effusions or locking of the knee.
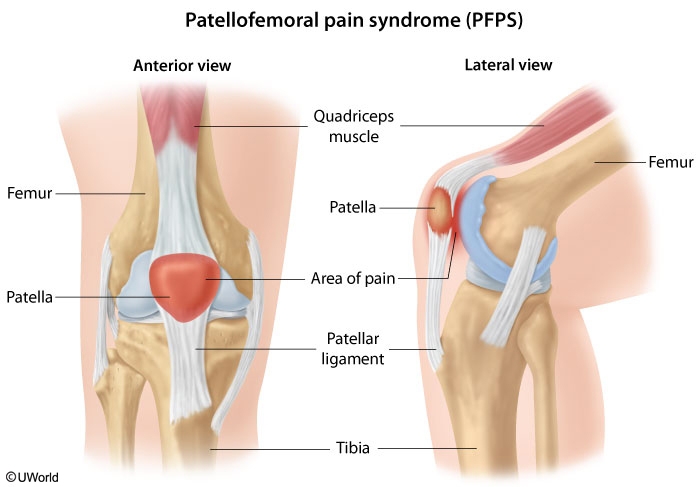
This patient with poorly localized anterior knee pain has typical features of patellofemoral pain syndrome (PFPS). PFPS is one of the most common causes of knee pain in young athletes and predominantly affects women. It is usually triggered by chronic overuse or malalignment (eg, angular deformities, weakness of hip abductors) but can also be seen acutely following trauma. The diagnosis is primarily based on clinical findings with pain localized to the anterior knee, usually described as an aching sensation, and worsened by activities such as climbing up or down stairs. The patellofemoral compression test (reproduction of pain when the patella is compressed into the trochlear groove) is often helpful, although no single finding has high sensitivity or specificity.
The initial management of PFPS includes reduced intensity of exercise (especially running), activity modification, and nonsteroidal anti-inflammatory drugs. Patients also should be counseled on stretching and strengthening exercises, with an emphasis on the quadriceps, knee extensors, and hip abductors.
Atlanto
Atlantoaxial instability is a malformation seen in 10%-15% of patients with Down syndrome, and most commonly occurs due to excessive laxity in the posterior transverse ligament, which causes increased mobility between the atlas (C1) and the axis (C2). Fortunately, only 1%-2% of Down syndrome patients with atlantoaxial instability are symptomatic. Symptoms usually progress over several weeks and result from compression of the spinal cord.
Presenting signs and symptoms include torticollis, urinary incontinence, and vertebrobasilar symptoms such as dizziness, vertigo, and diplopia, all of which may manifest as behavioral changes (as in this patient who is refusing to do usual activities). On examination, upper motor neuron findings such as leg spasticity, hyperreflexia, a Babinski sign, and clonus are often present. Patients with Down syndrome are normally hypotonic, and they may remain hypotonic or have increased tone with symptomatic atlantoaxial instability.
Atlantoaxial instability is suspected on physical examination and diagnosed with lateral radiographs of the cervical spine in flexion, extension, and in a neutral position. Open mouth radiographs can also be helpful in visualizing the odontoid. Treatment consists of surgical fusion of the first cervical vertebrae (C1) to the second (C2).
Croup

Croup, or laryngotracheitis, is a viral respiratory illness most commonly caused by parainfluenza virus and typically presents in children age 3-36 months. The illness usually begins with nonspecific symptoms (eg, rhinorrhea, congestion, fever); classic croup then presents with a dry, "barky," seal-like cough, hoarseness, and inspiratory stridor due to upper airway obstruction. The stridor worsens with agitation or excitement and may be inspiratory or biphasic (inspiratory and expiratory) in very severe cases.
Croup is typically a clinical diagnosis. If the diagnosis is unclear, anteroposterior neck radiographs will reveal subglottic edema known as the "steeple sign" (red arrow). Treatment is aimed at reducing subglottic edema; corticosteroids (eg, dexamethasone) are useful for mild cases and nebulized racemic epinephrine is added for patients with stridor at rest.

Bacterial sinusitis
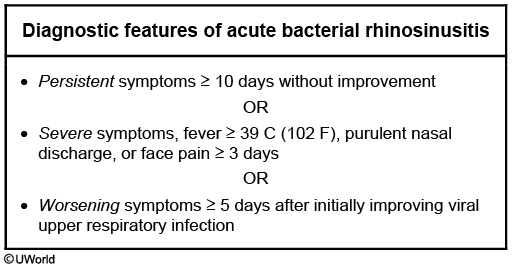
The duration of this patient's symptoms is consistent with acute bacterial rhinosinusitis. Streptococcus pneumoniae (30%), nontypeable Haemophilus influenzae (30%), and Moraxella catarrhalis (10%) are the most commonly implicated organisms. Due to increasing rates of beta-lactamase resistance, the treatment of choice is amoxicillin-clavulanic acid. Patients with asthma may experience exacerbations (eg, wheezing, coughing) triggered by concurrent upper respiratory infection.
Beckwith-Wiedmann

Beckwith-Wiedemann syndrome (BWS) is an overgrowth disorder characterized by a predisposition to neoplasms. Most patients have a sporadic or inherited alteration of chromosome 11p15, which includes genes that encode insulin-like growth factor 2, a growth-promoting hormone similar to insulin. At birth, classic physical findings include macrosomia, macroglossia, hemihyperplasia, and medial abdominal wall defects (umbilical hernia, omphalocele). Some infants also have visceromegaly.
Newborns must be monitored closely for hypoglycemia. Fetal hyperinsulinemia can result in profound hypoglycemia at birth (similar to infants of diabetic mothers). This problem is usually transient, and older asymptomatic patients usually do not require ongoing glucose monitoring (Choice D). Patients with BWS are at significantly increased risk of Wilms tumor and hepatoblastoma. Screening abdominal ultrasound and α-fetoprotein levels should occur every 3 months from birth to age 4 years, abdominal ultrasound every 3 months from age 4-8 years, and then renal ultrasound from age 8 years through adolescence.
Patients with isolated hemihyperplasia are also at increased risk for Wilms tumor and hepatoblastoma. These patients should undergo frequent screening as in BWS.
Digeorge
CATCH 22
conotruncal heart, abnormal faces, thymus aplasia, cleft palate, hypocalcemia
Cervical Lymphadenitis
Cervical lymphadenopathy is common in children. Lymphadenitis is diagnosed when the lymph node becomes tender and erythematous in addition to being enlarged. Although there are multiple causes for lymphadenopathy in children, the differential diagnosis can be narrowed by determining if the lymphadenopathy is acute or subacute/chronic, and if it is unilateral or bilateral. Acute, unilateral lymphadenitis in children is usually caused by bacterial infection. Staphylococcus aureus is the most common pathogen isolated, followed by group A streptococcus. Patients with bacterial lymphadenitis are usually less than 5 years old and nontoxic appearing. The affected lymph node is tender, warm, erythematous, and usually 3 to 6 cm in size. In some cases, the infection can progress to induration and fluctuance.
Peptostreptococcus is an anaerobic bacteria that can cause acute, unilateral lymphadenitis. However, it is usually seen in older children with a history of periodontal disease.
Nontuberculous mycobacteria (most commonly Mycobacterium avium-intracellulare) are one cause of unilateral subacute-chronic lymphadenopathy. Affected children are usually less than 5 years old and present with firm, nontender lymphadenopathy that is usually less than 4 cm in size. The skin over the lymph node often thins and develops a violaceous color. Fever and tenderness are unusual with this infection.
Eosinophilic Esophagitis
This patient has abdominal pain that is related to eating and that does not respond to acid suppression (eg, proton-pump inhibitor), which is consistent with eosinophilic esophagitis. Symptoms are due to esophageal inflammation triggered by food allergens, and patients frequently have eczema (as seen in this patient) or other allergic conditions (eg, asthma, rhinitis). Presentation typically includes dysphagia (ie, difficulty swallowing), mid-chest and epigastric pain, vomiting, and food impaction. Food refusal or a preference for soft foods is common in children. These symptoms can lead to weight loss.
In patients in whom eosinophilic esophagitis is suspected, a 2-month trial of proton-pump inhibitor therapy is considered part of the diagnostic evaluation; if there is no symptom improvement following this trial, endoscopy with esophageal biopsy is performed. Circular rings and esophageal furrows are nonspecific findings that may be present on endoscopy; diagnosis is confirmed by ≥15 eosinophils per high-power field on histology. First-line treatment is dietary modification to avoid potential food triggers. Pharmacologic management can include topical (eg, swallowed) fluticasone.
Cefotaxime
Third-generation cephalosporins (eg, ceftriaxone or cefotaxime) are effective against most strains of S pneumoniae and N meningitidis; vancomycin is given due to increasing prevalence of resistant strains of S pneumoniae. In neonates (age <28 days), who often have physiologic hyperbilirubinemia, cefotaxime should be used as ceftriaxone displaces bilirubin from albumin and increases the risk of kernicterus.
Osteomyelitis
Osteomyelitis in children
Patient population
Most common organisms
Empiric antibiotic therapy
Healthy children
Staphylococcus aureus
Low likelihood of MRSA Nafcillin/oxacillin OR cefazolin High likelihood of MRSA Clindamycin OR vancomycin
Children with sickle cell disease
Salmonella spp Staphylococcus aureus
As above PLUS Third-generation cephalosporin (ceftriaxone, cefotaxime)
Group B Streptococcus and Escherichia coli are common causes of osteomyelitis and septic arthritis in infants age <2 months but are extremely uncommon causes in older children.
Marfan
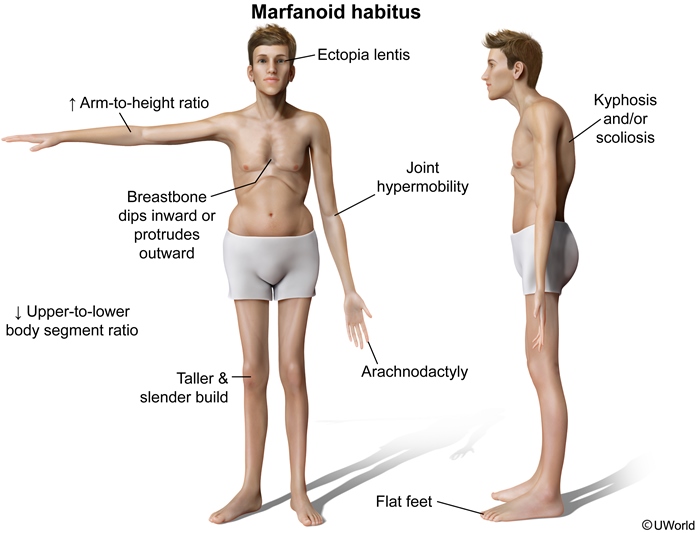
This patient's family history and physical appearance are very characteristic of Marfan syndrome. Marfan syndrome is an autosomal dominant disorder of the fibrillin-1 gene that results in systemic weakening of connective tissue. Classic skeletal manifestations include joint hypermobility, skin hyperelasticity, long fingers (arachnodactyly ["thumb sign"]), pectus excavatum, and scoliosis/kyphosis. The face is long, the palate has a high arch, and the teeth are crowded. Lens dislocation (ectopia lentis), iridodonesis (a rapid contraction and dilation of the iris), and myopia (from elongation of the globe) are also typical.
The most life-threatening finding in Marfan syndrome is aortic root dilation. The diastolic murmur in this patient reflects aortic regurgitation. The syndrome requires close monitoring with echocardiography for the development of aneurysms and aortic arch dissection. Mitral valve prolapse is also common and manifests as a mid-systolic click and late systolic murmur. First-degree relatives should undergo genetic testing.

Homocystinuria
Homocystinuria is an autosomal recessive disorder that results from deficiency of cystathionine synthase, an enzyme involved in the metabolism of methionine. These patients share many features of Marfan syndrome (eg, pectus deformity, tall stature, arachnodactyly). However, they usually have a fair complexion, thromboembolic events, and intellectual disability. The other main differentiating feature is lens dislocation in homocystinuria that is downward rather than upward.
Ehlers Danlos
Ehlers-Danlos syndrome is a collagen disorder characterized by scoliosis, joint laxity, and aortic dilation. Patients with this disorder do not have the disproportionately tall stature, lens dislocation, or pectus carinatum seen in Marfan syndrome.
Wilims

Wilms tumor (nephroblastoma) is the most common primary renal neoplasm of childhood. It is usually diagnosed at age 2-5 years and affects a single kidney. The most common presentation is an asymptomatic abdominal mass that is found incidentally by a caretaker or physician. Some patients have abdominal pain, hypertension, hematuria, and fever. Less than 10% of patients have bilateral renal involvement (stage V disease). Although the lungs are the most common site of metastatic spread, children rarely present with pulmonary symptoms.
Abdominal ultrasonography should be the first step in imaging to differentiate Wilms tumor from other causes of abdominal masses. It should be followed by contrast-enhanced computed tomography of the abdomen to evaluate the nature and extent of the mass and of the chest to identify any pulmonary metastases. Treatment includes surgery and chemotherapy with the addition of radiation therapy for high-stage disease. Survival rates are excellent especially if treated in the early stages.

Neuroblastoma can arise anywhere in the sympathetic nervous system but typically involves the adrenal glands and presents as an abdominal mass that crosses the midline with systemic symptoms. This patient's age and asymptomatic abdominal mass are more characteristic of Wilms tumor than neuroblastoma.
Hyposthenuria
This patient has a low urine specific gravity consistent with hyposthenuria, the inability of the kidneys to concentrate urine. Sickle cell trait (SCT) is the most likely cause of this patient's hyposthenuria.
Hyposthenuria is common in patients with SCD and may also develop in those with SCT. In response to hypoxic, hyperosmolar conditions of the renal medulla, red blood cells sickle in the vasa recta, impairing free water reabsorption and countercurrent exchange. Patients typically have polyuria and nocturia despite fluid restriction. Urine osmolality is low; however, normal serum sodium is maintained due to intact antidiuretic hormone (ADH). Urinary diluting capacity is also intact as it is a function of the superficial loop of Henle, which is not supplied by the vasa recta.
Typically, mild hyposthenuria due to SCT requires no treatment. In patients with SCD, red blood cell transfusions often improve urine-concentrating ability and provide relief of symptoms.
Murmur

Reye Syndrome
Presenting signs include vomiting and abnormal behavior, which progress rapidly to seizures and lethargy. Characteristic laboratory findings of hepatic injury include marked elevations in aminotransferases, hyperammonemia, and prolonged PT. Hypoglycemia may result from depletion of glucose stores and increased use. Despite the degree of liver involvement, total bilirubin is usually normal or minimally elevated, and icterus is unusual. Treatment is supportive. Elevated intracranial pressure is a major cause of death in Reye syndrome.
Patau
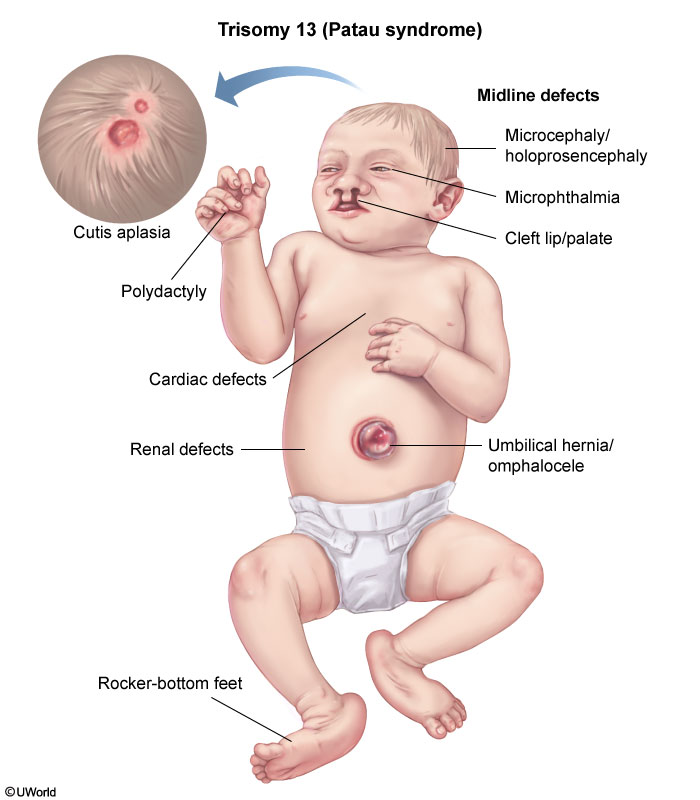
Cutis aplasia (absence of epidermis over the skull) and microphthalmia are both classically seen in trisomy 13 (Patau syndrome). This condition also associated with other midline defects, including holoprosencephaly and omphalocele.
Muscular Dystrophies

Myotonia (delayed muscle relaxation) is most notable when the patient is unable to release the hand after a handshake (grip myotonia). Skeletal muscle weakness is prominent in the face, forearms, hands, and ankle dorsiflexors (eg, bilateral foot drop). Dysphagia, the most dangerous smooth muscle manifestation, significantly increases the risk of aspiration pneumonia. Cardiac involvement includes conduction problems and arrhythmias. Other manifestations include cataracts, testicular atrophy/infertility, frontal baldness, and insulin resistance.
Achondroplasia

Achondroplasia is caused by a fibroblast growth factor receptor 3 (FGFR3) gene mutation that inhibits endochondral bone formation, primarily in long bones. Therefore, patients have disproportionate short stature (ie, shortening of the extremities relative to the trunk). Common dysmorphic features include frontal bossing and macrocephaly. Fractures and hearing loss are not typical.
Pyloric Stenosis
When the diagnosis is suspected based on clinical presentation, the best next step in management is abdominal ultrasonography. The presence of a thick and elongated pylorus confirms the diagnosis.

Homocysteinuria

Homocystinuria is an autosomal recessive disorder that results from errors in methionine metabolism secondary to cystathionine synthase deficiency. Patients with homocystinuria have a Marfanoid body habitus, including tall stature; long, thin limbs; joint hyperlaxity; ectopia lentis; and chest deformities. However, several clinical features are seen only in homocystinuria (Table). This child's fair hair and eyes, developmental delay, and cerebrovascular accident are virtually pathognomonic for homocystinuria. These patients may have thromboembolic events at any age due to pathologic changes in the vessel walls and increased adhesiveness of the platelets. Such events most commonly involve the cerebral vessels.
The diagnosis of homocystinuria is made based on elevated homocysteine and methionine levels. Treatment includes vitamin B6, folate, and vitamin B12 to lower homocysteine levels. In addition, antiplatelets or anticoagulation should be administered to prevent stroke, coronary heart disease, and venous thromboembolic disease.
Rheumatic Fever

This patient's history and examination findings are suggestive of rheumatic fever with persistent valvular disease (mitral stenosis with loud first heart sound and mid-diastolic rumble). Mitral stenosis almost always is due to rheumatic fever. Rheumatic fever occurs as a complication of pharyngeal infection with group A beta-hemolytic Streptococcus (GAS). All patients with an initial diagnosis of rheumatic fever should be treated with antibiotic therapy to eradicate GAS regardless of the presence or absence of pharyngitis at the time of diagnosis.
Patients with a history of rheumatic fever are at high risk for recurrence and progression of rheumatic heart disease with repeated episodes of GAS pharyngitis. All such patients should receive continuous antibiotic prophylaxis to prevent recurrent GAS pharyngitis. The preferred regimen is administration of intramuscular benzathine penicillin G every 4 weeks. The total duration of antibiotic prophylaxis depends on the severity of the disease (Table).
Cox vs Herpes

Differentiating between herpangina and herpetic gingivostomatitis can be challenging as both present with fever, pharyngitis, and oral lesions in young children. Herpangina is caused by the coxsackie A virus while herpetic gingivostomatitis is caused by primary infection with herpes simplex virus (HSV) type 1. Unlike HSV gingivostomatitis, herpangina has a seasonal pattern and infection most commonly occurs during summer/early fall.
The primary distinguishing feature between these illnesses is the location of the lesions. Herpangina typically presents with 1-mm gray vesicles on the tonsillar pillars and posterior oropharynx that progress to fibrin-coated ulcerations. It can be accompanied by lesions on the hands and feet (ie, hand-foot-mouth syndrome). In contrast, the vesicles in HSV gingivostomatitis (Choice D) generally localize to the anterior oropharynx and lips.
This patient's clinical presentation in the summer (prior to school entry) is consistent with herpangina. Treatment is supportive (eg, oral hydration and analgesia) as lesions self-resolve within 1 week.

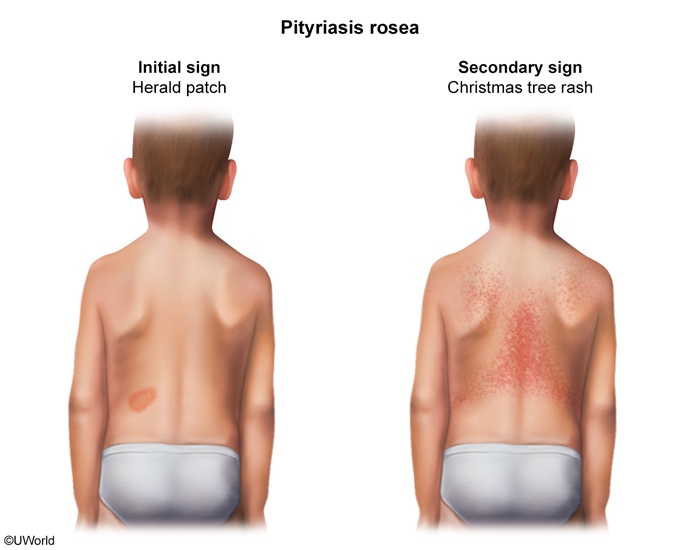
Pityriasis
Pityriasis rosea
Clinical features
± Viral prodrome Annular, pink herald patch on trunk Oval lesions in "Christmas tree" pattern Pruritus
Management
Reassurance (spontaneous resolution) Treatment of pruritus (eg, antihistamines)
Pityriasis rosea is a common skin condition that typically presents from early adolescence to young adulthood. The rash classically begins with a herald patch, an erythematous, annular lesion on the trunk. This patch may increase in size and develop scaling around the edge. Within a week, clusters of smaller, erythematous, oval lesions appear on the trunk. These scaly macules and papules are typically distributed obliquely along the lines of tension in a "Christmas tree" pattern, most noticeable on the back. The rash is often asymptomatic but may be associated with mild pruritus. A viral prodrome may precede pityriasis rosea, as seen in this patient with preceding fever, headache, and malaise.
Pityriasis rosea is self-limited and spontaneously resolves within weeks to months. Management is reassurance alone, although symptomatic relief of pruritus (eg, antihistamines, topical corticosteroids) may be indicated.
Emergency Contraception
copper IUD
ulipristal
oral lenonorgesterol
Lead toxicity
fingerstick capillary specimen
confirm with venous measurement
Chelation therapy is not routinely administered for lead levels <45 µg/dL due to lack of evidence for improved neurologic outcomes compared with removal from the lead-containing environment. Dimercaptosuccinic acid (succimer) is typically used when lead levels are 45-69 µg/dL. Dimercaprol (British anti-Lewisite) plus calcium disodium edetate (EDTA) should be administered on an emergency basis for levels ≥70 µg/dL or acute encephalopathy.
Bone Tumor Location

Ewing
Imaging of Ewing sarcoma classically reveals a lamellated periosteal reaction in which a central, poorly defined lytic lesion is surrounded by concentric layers of new bone ("onion skinning"). A "moth-eaten" or mottled appearance can also occur with extension into soft tissue. Codman triangle is a nonspecific radiographic finding that represents the displacement/elevation of periosteum by underlying tumor and can also be seen in osteosarcoma. Treatment includes chemotherapy as well as surgical resection ± radiation.
Central lytic lesion
"Onion skinning" (lamellated periosteal reaction)
"Moth-eaten" appearance
Periosteal elevation (Codman triangle)
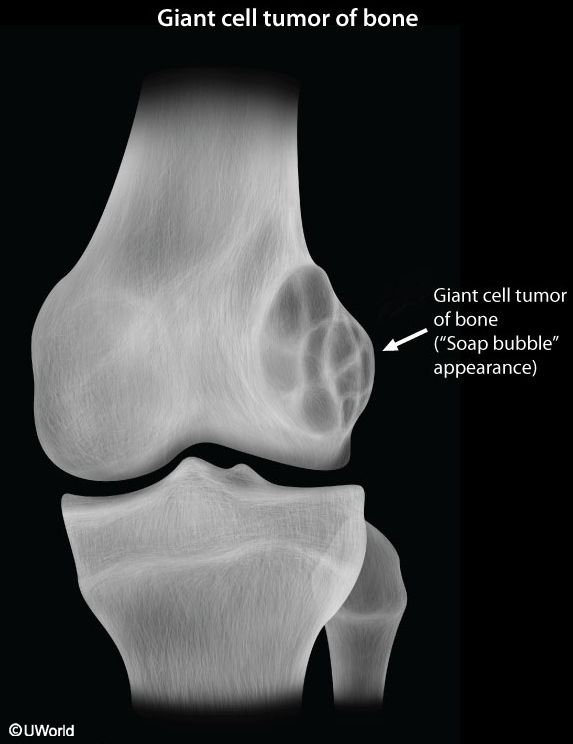
Multiple Myeloma
Extensive, punched-out lytic lesions, severe osteopenia, and pathologic fractures are common x-ray findings.

Cerebral Palsy
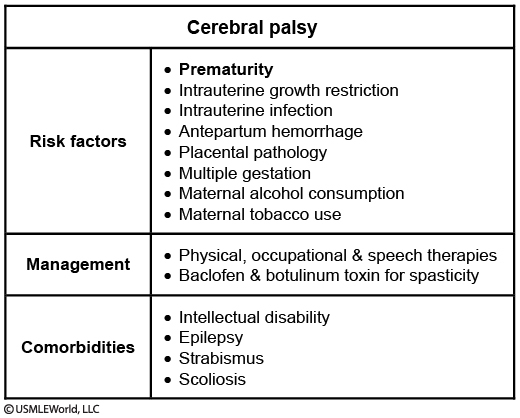
Cerebral palsy (CP) is a group of clinical syndromes characterized primarily by nonprogressive motor dysfunction. The 3 primary subtypes - spastic, dyskinetic, and ataxic - are often multifactorial in etiology. CP is usually caused by prenatal insults to brain development, with premature birth before 32 weeks gestation as the greatest risk factor. Spastic diplegia is the form most commonly seen in preterm infants. It presents as hypertonia and hyperreflexia that involve predominantly the lower extremities with both feet pointing down and inward (equinovarus deformity). Resistance to passive muscle movement increases with more rapid movement of the affected extremity ("clasp-knife").
Many patients with CP suffer from vision, hearing, speech, or other impairments (Table). Approximately 50% of patients also have intellectual disability. Management involves multidisciplinary therapies and anti-spastic medications to prevent and improve contractures.
Volvulus

The finding of the Ligament of Treitz on the right side of the abdomen reflects malrotation while contrast in a "corkscrew" pattern indicates volvulus.
Last updated