01 Cardiology
General
Cardiac Anatomy
The coronary arteries originate from the aortic sinus, the region of the aorta just above the aortic valve.
The right coronary artery (RCA) supplies the right ventricle, as well as theposterior portion of the interventricular septum in most people. When the RCA supplies the posterior heart, this is known as right dominance.
The right coronary artery (RCA) passes anteriorly, to the right of pulmonary artery root, coursing posteriorly through the sulcus between the right atrium and right ventricle.
The sinoatrial artery originates from the right coronary artery (RCA) to supply the sinoatrial node. Acute thrombus in the proximal RCA may result in the failure to generate regular sinus rhythm.
The right marginal artery originates from the right coronary artery (RCA), and serves as the main supply to the right ventricle.
In most people, the right coronary artery(RCA) also gives rise to the posterior descending artery (PDA), which courses between the two ventricles posteriorly and supplies the posterior region of the interventricular septum, as well as some of the posterior right and left ventricles. When the RCA gives rise to the PDA (as it does in 80% of people), this is known as right dominance.
The two main branches of the left coronary artery (LCA, sometimes 'LMCA' for left main coronary artery) are the left anterior descending artery (LAD) and the left circumflex artery (LCx).
The left coronary artery (LCA) passes anteriorly, to the left of the pulmonary artery root, then divides into its two major branches about halfway around the left atrium. Because the course of the LCA is so short, it is typically not considered to have its own proper supply domains.
The left anterior descending artery (LAD) descends between the right and left ventricle to supply the anterior interventricular septum, the more left aspects of the right ventricle, and the majority of the left ventricle.
The left circumflex artery (LCx) courses posteriorly between the left atrium and left ventricle to supply the more posterior aspects of the left ventricle.
In some people (20%), the left circumflex artery (LCx) gives rise to the posterior descending artery (PDA), which supplies theposterior region of the interventricular septum as well as some of the posterior right and left ventricles. This is known as left dominance.
Bloodflow through the coronary arteries is biphasic, with greater flow occurring during diastole.
The systolic ventricular pressure is far greater on the left than on the right, such that the left ventricle is essentially perfused during diastole only.
Cardiac Physiology
Cardiac output (CO) is defined as the _product_of the stroke volume (SV) and the heart rate (HR): CO = SV x HR
Stroke volume is the amount of blood ejected from a ventricle during systole. With respect to the left ventricle, this is equal to the difference between left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV): SV=LVEDV−LVESVSV = LVEDV - LVESVSV=LVEDV−LVESV.
A decrease in stroke volume should produce a compensatory increase in heart rate in an effort to maintain cardiac output, as occurs in the settings of hypovolemic shockand (sometimes) distributive shock.
Mean arterial pressure (MAP) is defined as the average arterial pressure over the cardiac cycle, and is derived from central venous pressure (CVP), cardiac output (CO), and systemic vascular resistance (SVR): MAP=(CO×SVR)+CVPMAP = (CO\times SVR)+CVPMAP=(CO×SVR)+CVP
Mean arterial pressure (MAP) for a normal person at rest can be estimated from the systolic (BPS) and diastolic (BPD) blood pressures, taking into account the relative contributions of each phase of the cardiac cycle: MAP≈13BPS+23BPDMAP \thickapprox \frac{1}{3}BP_S + \frac{2}{3}BP_DMAP≈31BPS+32BPD. (MD calc provides an easy way to do this on the wards.)
Since MAP accounts for arterial pressure during systole and diastole, it is considered the most physiologic measurement of the pressure perfusing the body’s organs.
Arterioles are primarily responsible for determining systemic vascular resistance in the absence of anatomic anomalies (e.g. arteriovenous malformations).
An adequate systolic arterial blood pressure is required to perfuse the body’s organs; accordingly, a decrease in systemic vascular resistance should produce a compensatory increase in cardiac output. This is primarily due to an increase in heart rate, and is observed in the setting of septic shock, a form of distributive shock.
CAD
CAD Overview
Coronary artery disease occurs when the coronary arteries become progressively narrowed and lose their ability to dilate, causing a mismatch between myocardial oxygen supply and demand. This is most commonly the result of extensive atherosclerotic plaque formation.
Symptomatic CAD is divided into stable angina and the acute coronary syndromes, including unstable angina, NSTEMI and STEMI; however, many patients with CAD are asymptomatic.
Heart disease as a broad etiologic category remains the leading cause of death in the United States at 253 per 100,000 people. Ischemic heart disease is the single most common diagnosed cause of death in the US at 123 per 100,000 people.
Coronary artery disease is strongly associated with major modifiable risk factors:
Tobacco usage
Hypertension
Sedentary lifestyle
Obesity
Diabetes mellitus
Coronary artery disease (CAD) is associated with the following major uncontrollable risk factors:
Age > 65
Sex: men are at higher risk
Family history: premature coronary disease in men under 55 and women under 65
Coronary artery disease begins as intimal fatty streaks early in life, but the plaques and thrombi that result in the clinical consequences develop later on in adulthood.
For diagnosis and treatment of CAD-related risk factors, see:
Atherosclerosis
Hypercholesterolemia
Primary Hypertension
Secondary Hypertension
For diagnosis and treatment of CAD-related conditions, see:
CAD: Stress tests
CAD: Stable angina pectoris
Acute Coronary Syndromes
CAD Diagnosis
The resting 12-lead electrocardiogram (ECG) is generally the initial diagnostic test used to evaluate ischemic heart disease, because it allows for the most physiologic evaluation of myocardial perfusion.
Most patients require stress testing for further diagnosis. The initial stress test could be any one of the following and should be based on resting ECG, patients ability to perform exercise, and available technology:
Exercise ECG with/without imaging
Pharmacologic ECG with imaging
Patients who cannot exercise can undergo pharmacologic stress testing, in which a drug replaces exercise as the stimulus for increasing myocardial perfusion. In pharmacologic stress testing, ECG is always combined with an imaging modality.
Dobutamine, a cardiac inotrope, is used in pharmacologic stress echocardiography to evaluate patients who cannot exercise.
The vasodilators adenosine and dipyridamole are used in radionuclide myocardial perfusion imaging (rMPI). Perfusion is restricted in territories supplied by stenotic vessels which are unable to dilate, so there is comparatively less radionuclide uptake in these regions.
The type of diagnostic testing that is appropriate for a given patient is dependent upon his or her pre-test probability of CAD . Determining the appropriate test for a given patient vignette is very high-yield for the USMLE Step 2.
The pre-test probability of coronary artery disease is described in the attached chart. As a reminder, the pre-test probability affects the false negative and false positive rates. Low probability patients are more likely to have a falsely positive test, while high probability patients are more likely to have a false negative. This is why patients with low pre-test probability for CAD are not screened.

Typical: substernal pain, increased with exersion, relieved by rest/nitrate
Atypical: 2 of 3
Nonanginal: 0 or 1
In general, diagnostic testing for CAD is warranted in patients with symptoms of CAD, asymptomatic patients with high pre-test CAD probability, or patients with newly diagnosed heart failure.
Asymptomatic
The only asymptomatic patients who should undergo stress testing are those with a global risk of >20% for clinical CAD within the next 10 years. They should undergo exercise ECG unless contraindicated due to inability to exercise. (There are several models discussed in the 2010 ACCF/AHA guideline for determining 10-year global risk for CAD, and this is generally beyond the scope of the USMLE exams--and thus of this product. One popular model is the Framingham risk score, which takes into account factors such as age, sex, total cholesterol, HDL cholesterol, smoking, systolic blood pressure, and treatment with antihypertensive medications. Other models factor in diabetes and family history.)
Symptomatic
Symptomatic patients with low or intermediate pre-test probability who are both 1) able to exercise and 2) have an interpretable ECG should undergo exercise stress testing.
Stress radionuclide imaging or echo are the tests-of-choice for most symptomatic patients who are unable to exercise or have an uninterpretable ECG. They may also be used for symptomatic intermediate and high pre-test probability patients.
Stress cardiac MRI is primarily for symptomatic high pre-test probability patients.
In other words, a symptomatic patient with high pre-test probability should receive stress radionuclide imaging, stress echo, stress MRI, or coronary angiography, NOT an exercise ECG.
CHF
Newly diagnosed CHF should be evaluated for coronary artery disease similarly to symptomatic patients with high pre-test probability: stress radionuclide imaging, stress echo, stress MRI, or coronary angiography, NOT an exercise ECG.
Exercise stress testing combines controlled exercise (e.g. treadmill) with an EKG to visualize ischemic changes or induce angina.
Exercise stress ECG is considered positive for ischemic coronary artery disease with any of the following results:
Greater or equal to 1 mm ST-segment depression which is horizontal or down-sloping
Greater than 10 mm Hg drop in systolic blood pressure (SBP) from resting value, OR failure to elevate SBP to > 120 mm Hg
Some drugs should be held for exercise stress tests because they will, by their nature, alter the EKG results:
Beta-blockers
Non-dihydropyridine calcium channel blockers (i.e. diltiazem, verapamil)
Certain antiarrhythmic agents (e.g. amiodarone, sotalol)
Digoxin
Nitrates
Exercise stress testing is contraindicated in unstable patients (e.g. hypertensive emergency, aortic dissection, acute myocardial infarction), in patients who cannot exercise (e.g. mobility restrictions) or patients with uninterpretable EKGs (e.g. patients with pacemakers, resting ST-segment changes, bundle branch blocks, or on digoxin.)
Low-grade coronary artery stenosis (less than 50%) isn’t severe enough to cause EKG changes, which means the exercise stress test will appear normal.
Nuclear stress testing uses a radioactive substance like thallium-201 or technetium-99m (Tc-99m) sestamibi to allow visualization of heart tissue perfusion by single photon emission CT scan during rest and exercise.
Thallium accumulates in well-perfused heart tissue during a nuclear stress test.
Nuclear stress tests are appropriate in patients with baseline EKG abnormalities (e.g. bundle branch block, left ventricular hypertrophy, or a pacemaker) because the results are not affected by these abnormalities.
Stress echocardiogram combines an exercise stress test with an echocardiogram, which enables recognition of heart wall motion abnormalities during exercise.
Coronary angiography is the most sensitive and specific for CAD, however, it is the most invasive. Unlike all other diagnostic tests, it allows for immediate intervention (e.g. stent placement).
See Acute Coronary Syndromes for more information.
STEMI Diagnosis
ST-elevation myocardial infarction (STEMI) is a type of acute coronary syndrome in which a patient presents with new (or presumed new) territorial ST-segment elevation on electrocardiogram (ECG).
Specific ECG diagnostic criteria for STEMI are:
> 1mm persistent ST-elevation in 2 or more contiguous leads, or
> 2mm persistent ST-elevation in leads V2 and V3, or
New left bundle-branch block (LBBB), or
True posterior MI (look for reciprocal ST depression in the anterior leads)
The EKG in a STEMI changes over time. It is important to recognize that these changes do not always occur in this classic sequence, and multiple findings may be simultaneously present.
Tall and peaked hyperacute T waves will be seen over the ischemic zone due to local hyperkalemia.
J point elevation with preservation of ST segment morphology
The ST segment develops convexity and becomes rounded upward, merging progressively with the T wave
Pathologic Q waves develop as myocardium dies.
In patients who do not achieve reperfusion, persistent T wave inversions develop in the region of the infarct, but the ST segment returns to baseline


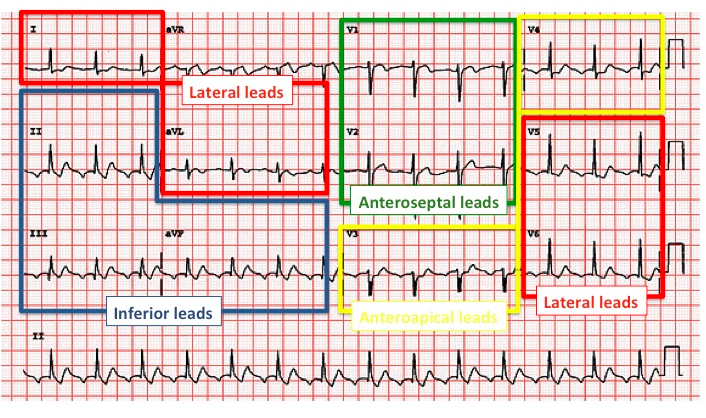
Territorial ST-segment elevation typically reflects transmural ischemia.
ST elevation in leads II, III, and aVF indicates an inferior infarct of the posterior descending artery or marginal branch.
ST elevation in leads I, aVL, and V5-V6 indicates a lateral infarct of the left anterior descending artery or circumflex.
ST elevation in leads V1-V2 indicates a septal infarct of the left anterior descending artery.
ST elevation in leads V3-V4 indicates anterior infarct of the left anterior descending artery.
Patients with inferior infarct should also be evaluated with a right-sided EKG for right-heart infarction. This is imperative because patients with right-sided infarction are preload dependent and cannot receive nitrates.
Serum cardiac enzymes, specifically cardiac-specific troponin T (cTnT), cardiac-specific troponin I (cTnI), and creatine kinase MB-isoenzyme (CKMB), are crucial for diagnosing and monitoring progression of a STEMI.
Troponins are more sensitive and specific for myocardial injury than creatine kinase (including the MB fraction.)
CKMB rises within 8 hours of infarction and returns to normal by 72 hours, but troponins remain elevated for 7-10 days after infarction and cannot be used to assess for reinfarction during this period.
In ruling out myocardial infarction, patients are typically admitted to have 3 separate sets of cardiac enzymes drawn, each 6-8 hours apart, as the overwhelming majority of patients with myocardial infarction will have positive enzymes by 16 hours.
STEMI Evaluation
ST-elevation myocardial infarction (STEMI) occurs when a thrombus abruptly occludes an atherosclerotic coronary artery, leading to transmural ischemia with irreversible myocardial injury.
Slowly developing coronary stenosis typically does not cause STEMI because a rich collateral circulation develops; however, this does predispose a patient to unstable angina and NSTEMI.
The most important risk factors for STEMI are having multiple atherosclerotic risk factors (e.g. smoking, hyperlipidemia) and unstable angina.
Less common but still important risk factors for STEMI include hypercoagulability, cocaine use, and collagen vascular diseases.
Initial pharmacologic treatment of STEMI is the same as unstable angina/NSTEMI (mnemonic: BEMOANS):
β-blocker
Unless heart failure, bradycardia, heart block, cardiogenic shock
Enoxaparin (reduces probability of recurrent coronary events)
Morphine (if in severe pain)
Oxygen (if SaO2<90% or dyspnea)
Anti-platelet: aspirin plus P2Y12 inhibitor (e.g. clopidogrel)
Nitrates (primary benefit from preload reduction; also reduces afterload)
Statin (e.g. high-dose atorvastatin)
Glucocorticoids and NSAIDs (except aspirin) are contraindicated in STEMI because they impair healing of the infarcted region and increase the risk for ventricular wall rupture.
The definitive treatment for STEMI is reperfusion therapy, which should be considered for every STEMI patient.
Fibrinolysis with tissue plasminogen activator (tPA), streptokinase, tenecteplase, or reteplase is referred to as “chemical reperfusion.”
Percutaneous coronary intervention (PCI) is a form of mechanical reperfusion therapy which can be performed along with diagnostic cardiac catheterization. PCI with stenting was shown to offer a significant reduction in reinfarction or stroke at 30 days, as well as significant mortality benefit for high risk patients with TIMI score > 5. [Nielson et al. Circulation. 2010;121:1484.]
Chemical fibrinolysis is indicated for the treatment of STEMI in patients presenting within 12 hours of the onset of symptoms when PCI cannot be accomplished within 120 minutes of first medical contact.
Primary PCI without antecedent fibrinolysis is preferred in the following situations:
Patients with absolute or otherwise compelling contraindications to fibrinolysis
When the diagnosis is in doubt
When there is cardiogenic shock
Absolute contraindications to chemical fibrinolysis are blood, blood, blood, BP, brain:
Cerebrovascular hemorrhage at any time
Active internal bleeding
Suspicion of aortic dissection
Marked hypertension (systolic >180 or diastolic >110) at any time during the acute presentation
Nonhemorrhagic stroke or other cerebrovascular event within the past year
Coronary artery bypass grafting (CABG) is indicated as the primary reperfusion strategy for the treatment of STEMI in patients with occlusion of the left main coronary artery or patients with severe three-vessel involvement.
Stable Angina
Stable angina pectoris is a subtype of coronary artery disease in which chest pain due to myocardial ischemia occurs predictably during periods of increased oxygen demand (e.g., exercise) or decreased supply (e.g., hypotension) and is relieved by rest or nitroglycerin.
Typical angina is substernal chest pain most commonly described as a squeezing, tightness, pressure sensation that is provoked by exertion or emotional stress and relieved by rest or nitroglycerin. Its onset and offset are gradual and repeat episodes typically present the same way.
Note that chest pain relieved by nitroglycerin is NOT entirely specific for angina--nitroglycerin may also soothe the pain associated with diffuse esophageal spasm.
Atypical angina refers to chest pain symptoms that are typically non-cardiac in nature:
Sharp, knife-like, pleuritic chest pain
Primary location in the mid-to-lower abdomen
Discomfort is easily localized (e.g. patient points with one finger to where it hurts)
Pain is associated with or worsened by movement or palpation
Pain is either fleeting or persistent
Patients with stable angina present with complaints of chest pain lasting between 5 and 15 minutes which may radiate to the jaw, neck, or shoulders, and was brought on by physical exertion. Cardiac pain that occurs at rest is more consistent with unstable angina.
Profuse sweating, shortness of breath, and a feeling of impending doom are more suggestive of myocardial infarction and are not typically part of the presentation of stable angina pectoris.
Atypical symptoms like weakness, breathlessness, nausea, vomiting, and midepigastric discomfort or sharp chest pain are considered “angina equivalent” in the elderly and women. Diabetics may also have an atypical presentation due to decreased sensitivity of sensory neurons involved in the generation of typical angina.
The physical exam of a patient suffering from stable angina pectoris is normal.
During an attack, or more frequently during deliberately induced high-oxygen-demand states like stress tests, electrocardiogram (ECG) will generally reveal regional ST-segment depression.
Ischemia-induced myocardial dysfunction may result in a paradoxically split S2; an S3 and/or an S4 may be present as well, which is caused by delayed relaxation of the left ventricular myocardium and delayed aortic valve closure.
The gold standard for diagnosing stable angina pectoris is the exercise stress test.
Stable angina pectoris is primarily treated with lifestyle modification and medical managment, though some patients may require revascularization.
Lifestyle modifications alter the major modifiable risk factors for coronary artery disease:
Smoking cessation
Blood pressure control
Increased activity level
Weight loss
Diabetes control
Beta-blockers are recommended as first-line agents for the reduction of anginal episodes and to improve exercise tolerance. Fast-acting nitrates provide relief from acute anginal symptoms; long-acting nitrates or calcium channel blockers may be used in combination with a beta-blocker if angina persists. Calcium channel blockers are also appropriate for patients who cannot tolerate beta-blocker therapy.
Aspirin and statins help reduce risk of acute thrombus, and should be used in stable angina patients.
UA and NSTEMI
Unstable angina (UA) is defined as chest or arm pressure or pain with at least one of three features: (1) it occurs at rest (or with minimal exertion) for >10 minutes; (2) it is severe and of new onset; and/or (3) it occurs with a crescendo pattern (i.e. more severe, longer, or more frequent episodes.) Unstable angina may also be called “crescendo angina.”
The most common cause of UA/NSTEMI is believed to be rupture of coronary artery plaques and subsequent down-stream occlusion.
In unstable angina, complete obstruction is rare; incomplete stenosis or presence of well-perfused collaterals may prevent progression to complete infarction (e.g. ST-elevating myocardial infarction).
Unstable angina and non-ST elevation myocardial infarction (NSTEMI) are, by definition, not associated with ST-elevation on EKG.
Some patients may present with ST-segment depression and/or T-wave inversion.
Territorial ST-segment depression typically represents a corresponding region of subendocardial (i.e. not transmural) ischemia. This occurs because the ischemic region is electrically depolarized even when the ventricle is at rest or repolarized, causing an elevation of the isoelectrical baseline. When the whole ventricle depolarizes, the ischemic region also depolarizes down to the same level. This gives the appearance that the ST segment is dropping below the isolectric baseline.
Unstable angina is not associated with changes in biomarkers for cardiac necrosis (troponins and CKMB).
NSTEMI is distinguished from unstable angina by the presence of elevations in cardiac enzymes. It is distinguished from STEMI by the absence of ST-segment elevation.
Patients who are elderly, have diabetes, or are women are more likely to present with atypical symptoms of ACS in the absence of chest pain, including:
Dyspnea
Weakness
Nausea, vomiting
Palpitations
All patients with suspected UA vs. NSTEMI should be admitted for telemetry and serial cardiac enzymes (i.e. at least two troponin measurements), as these are part of the TIMI score used to predict level of ACS risk and determine whether a patient may benefit from early coronary angiography and revascularization.
Immediate angiography and revascularization is recommended for patients with non-ST elevation ACS and at least one of the following:
Hemodynamic compromise or cardiogenic shock
Systolic heart failure
Recurrent or persistent angina despite medical therapy
Evolving mitral insufficiency or ventricular septal defect
Sustained ventricular arrhythmias
Initial pharmacologic treatment of UA/NSTEMI is the same as STEMI with the singular exception that fibrinolytics are never indicated. Otherwise, pharmacologic therapy includes (mnemonic: BEMOANS):
β-blocker
Unless heart failure, bradycardia, heart block, cardiogenic shock
Enoxaparin (reduces probability of recurrent coronary events)
Morphine (if in severe pain)
Oxygen (if SaO2<90% or dyspnea)
Anti-platelet: aspirin plus P2Y12 inhibitor (e.g. clopidogrel)
Nitrates (primary benefit from preload reduction; also reduces afterload)
Statin (e.g. high-dose atorvastatin)
The Thrombolysis In Myocardial Infarction (TIMI) Risk Score allows for grading risk of serious adverse outcomes in UA/NSTEMI, which is slightly different than the TIMI risk score used for STEMI. If TIMI risk score ≥ 3, consider early low molecular weight heparin and angiography.
The components of the TIMI score for UA or NSTEMI are (1 point each) SCAARES:
Severe angina (≥2 events over the past 24 hours)
Coronary artery stenosis ≥50%
Age ≥65 years
Aspirin use within the past 7 days
Three or more Risk factors for CAD (Fam history, DM, HTN, Smoking, dyslipidemia)
Enzymes: elevated troponins or CKMB
ST segment changes on ECG
Prinzmetal Angina
Vasospastic angina (previously known as Prinzmetal or variant angina) is anginal chest pain caused by coronary vasospasm.
Cigarette smoking is the primary cardiovascular risk factor for vasospastic angina.
In contrast to angina pectoris, vasospastic angina is not associated with other cardiovascular risk factors such as hypertension and hypercholesterolemia.
Coronary artery spasm in vasospastic angina most commonly occurs at sites of atherosclerotic plaques, although it can also occur in normal vessels.
Vasospastic angina may be associated with other vasospastic conditions, particularly Raynaud phenomenon and migraine headache.
Vasospastic angina presents more commonly in younger patients without a cardiac history with seemingly random episodes of severe resting chest pain that occur most frequently at night.
During an episode of chest pain, patients will have ST segment elevation on electrocardiogram (ECG), followed by a return to a normal ECG as the vasospasm and chest pain relent.
ST-elevation is caused in this setting by transmural ischemia in the absence of collateral circulation.
There is a high prevalence of severe life-threatening arrhythmias (e.g. ventricular fibrillation, ventricular tachycardia, or severe heart blocks) in variant angina, which is attributed to sudden coronary reperfusion.
Coronary angiography with a provocative test for vasospasm (e.g. ergonovine or acetylcholine infusion directly into the vessel) is the gold standard both for diagnosis and responsiveness to therapy.
Calcium-channel blockers (e.g. diltiazem) are the first-line treatment for vasospastic angina.
The pain of vasospastic angina is relieved by nitroglycerin.
Aspirin and nonselective beta blockers (e.g. propranolol) will worsen symptoms of vasospastic angina and should be avoided.
Since sumatriptan is associated with coronary vasospasm, it should be avoided in patients with vasospastic angina.
Recall that vasospastic angina is associated with other vasospastic disorders such as migraine headaches, so avoiding triptans in these patients is particularly relevant.
Post MI
The most common complication following myocardial infarction (MI) is arrhythmia.
Arrhythmias as a complication of MI typically occur during or immediately following the event.
Ventricular fibrillation is the most common cause of death in patients with or recently recovering from MI.
Infarction of parts of the interventricular septum may present with new onset atrioventricular or bundle branch blocks.
Structural complications of MI include ruptures and aneurysm.
The most common ruptures that occur as complications of myocardial infarction are:
Ventricular free wall rupture, resulting in acute pericardial tamponade
Ventricular septal rupture, resulting in a new ventricular septal defect murmur
Papillary muscle rupture, resulting in severe mitral regurgitation and hemodynamic collapse
**Note that the likelihood of any one of these occurring is predicted by the electrocardiogram at MI presentation.
Ruptures as a complication of MI generally occur 3 to 14 days following the event, once macrophages have cleared the debris.
Aneurysm
Aneurysms occur as a late (days to months) consequence of transmural infarcts that result in a large area of noncontractile transmural scar on a free wall which progressively dilates.
ECG findings suggestive of a left ventricular aneurysm include persistent ST elevation following a recent myocardial infarction and deep Q waves.
Sequelae of a left ventricular aneurysm include:
Cardiogenic shock
Arrhythmias
Thromboembolism (aneurysms are commonly filled with a clot)
The wall of a ventricular aneurysm paradoxically bulges outward during systole; this systolic bulge can be palpated on physical exam on the chest wall.
Pseudoaneurysm
Ventricular pseudoaneurysm is a complication of a MI that occurs when myocardial rupture is contained by pericardial or granulation tissue. In contrast to a true ventricular aneurysm, ventricular pseudoaneurysms are likely to rupture, leading to a potentially fatal hemorrhage.
Patients with left ventricular pseudoaneurysm are managed with emergency surgery since untreated pseudoaneurysms are prone to rupture.
Percarditis
The two inflammatory complications of MI include peri-infarction pericarditis (PIP) and Dressler's syndrome (postcardiac injury syndrome).
PIP is an acute inflammatory process extending to the pericardium that occurs 2 to 3 days following transmural MI as a normal response to clear necrotic tissue.
PIP presents with a pericardial friction rub with or without chest discomfort 2 to 3 days following MI.
The treatment of PIP is supportive, as most cases are self limited.
Dressler
Dressler's syndrome is an inflammatory response to previously sequestered pericardial antigens that develops 2-3 weeks following acute transmural MI.
Dressler's syndrome presents with low-grade fever, chest pain, pericarditis as evidenced by the presence of a friction rub on cardiac auscultation, and/or pericardial effusion. The time period will distinguish it from peri-infarction pericarditis (PIP).
Dressler’s syndrome is usually self-limited, but can be treated with aspirin or ibuprofen; prophylaxis may be provided with colchicine in the setting of cardiac surgery, but is not commonly used for MI patients.
MI long term management
Long-term management of patients who have suffered a myocardial infarction focuses on reducing risk of re-infarction and pathologic hypertrophy.
Important lifestyle modifications include smoking cessation, exercise, and dietary modifications targeted to decreasing weight, blood pressure, and cholesterol.
Important pharmacologic management BAA(N)S a second MI:
β-blocker
Low-dose Aspirin
ACE-inhibitor or ARB
Nitroglycerin (as-needed for recurrent angina)
Statin
**Note that nitroglycerin provides symptomatic relief only, while the other drugs reduce adverse remodeling and lower the risk for recurrent MI. These medications should be started before the patient leaves the hospital.
Patients who have received coronary interventions (e.g. PCI) should be loaded with aspirin and a platelet ADP receptor (P2Y12) blocker such as clopidogrel, ticagrelor, or prasugrel as soon as the diagnosis of STEMI is made, and should be discharged on a maintenance dose of these two antiplatelet medications.
The use of GpIIb/IIIa inhibitors is not routinely recommended for patients undergoing PCI for STEMI.
Chronic Heart Disease
Primary HTN
Primary (essential) hypertension is hypertension which occurs in the absence of an identifiable underlying etiology (i.e. idiopathic). It accounts for more than 95% of hypertension.
There are quite a few risk factors for the development of essential hypertension:
Alcohol consumption
Tobacco use
Depression
Excess dietary salt
Obesity
Geriatric population
Family history
Insulin-dependent diabetes mellitus
Blacks generally have a higher salt-sensitivity than whites, which contributes to a greater susceptibility to developing primary hypertension.
Primary hypertension is asymptomatic for many years. Headache may be the only symptom until complications develop.
Per 2017 AHA Hypertension Guidelines, primary hypertension is diagnosed if the blood pressure is determined on at least two separate occasions to be ≥130 mmHg systolic pressure and/or ≥80 mmHg diastolic pressure, and once common contributing factors have satisfactorily been ruled out.
At minimum, the following labs should be ordered in the workup of newly-diagnosed hypertension:
Blood chemistries (electrolytes, glucose, creatinine)
Urinalysis
Lipid profile
Electrocardiogram
Physical exam of the patient with identified essential hypertension should specifically look for evidence of end-organ damage.
Fundoscopic signs of end-organ damage include:
Arteriovenous nicking (apparent retinal-vein narrowing secondary to arterial wall thickening)
Cotton-wool spots
Retinal flame hemorrhages
Auscultation of the heart may reveal a loud S2 and classically a S4 component ('Tennessee gallop'), though a S3 may be present as well or instead. A left ventricular heave may indicate left ventricular hypertrophy.
Untreated or poorly treated hypertension increases the risk of:
Coronary artery disease (CAD)
Stroke
Aortic dissection
Congestive heart failure
Kidney disease
Vision impairment
Lifestyle modifications are the initial treatment for primary hypertension. In order of most to least effective, these include:
Weight loss (in obese patients)
DASH diet
Exercise
Salt restriction
Reduced alcohol consumption
Note that on NBME examinations, the cheapest option that is still an acceptable form of accomplishing the indicated step in management is USUALLY the correct answer.
According to the Joint National Committee (JNC 8), any of the following are an appropriate initial drug of choice for the treatment of primary hypertension:
Thiazide diuretic (e.g. hydrochlorothiazide, chlorthalidone)
Ca2+ channel blocker (e.g. amlodipine)
Angiotensin-converting enzyme(ACE) inhibitors (e.g. lisinopril)
Angiotensin II receptor blockers (ARBs) (e.g. losartan)
Patients with history of proteinuria in the setting of chronic kidney disease should receive an ACE inhibitor or angiotensin receptor blocker (ARB) as initial monotherapy.
Once a medication has been initiated, it should be titrated to achieve the goal blood pressure (i.e. patient is no longer hypertensive) up to the maximum dose before adding a second medication.
Black patients on ACE inhibitor monotherapy historically have had smaller blood pressure reductions compared to white patients. Therefore, ACE inhibitors should be avoided as the initial drug of choice. ACE inhibitors may be added for combination therapy or if monotherapy is insufficient.
If a patient's hypertension is not controlled on the maximum dosage of the initial drug, a second drug from a different class (i.e. calcium channel blocker, ACE inhibitor or ARB, thiazide diuretic) should be added and titrated to effect. Note that an ACE inhibitor and an ARB count as the same drug class for the purpose of hypertensive pharmacotherapy.
If a patient's hypertension is not controlled on the maximum dosages of two drugs, a third drug from a different class (i.e. calcium channel blocker, ACE inhibitor or ARB, thiazide diuretic) should be added and titrated to effect. Note that an ACE inhibitor and an ARB count as the same drug class for the purpose of hypertensive pharmacotherapy.
If a patient's hypertension is not controlled on the maximum dosages of three drugs, additional antihypertensive agents are indicated, and the patient requires referral to a hypertensive specialist. Note that when a patient's hypertension is resistant to triple therapy there is very likely an underlying etiology present, such as renal artery stenosis.
Hypotension
Hypotension is abnormally low blood pressure which is a pathological deviation from an individual's physiologic baseline, but is generally defined as a systolic blood pressure < 90 mmHg or a diastolic blood pressure <60 mmHg.
Individuals who regularly engage in intense physical training may normally exhibit a low blood pressure at rest.
Several common causes of hypotension to consider include:
Hypovolemia, which may be due to hemorrhage, increased fluid loss (e.g. diarrhea, vomiting), increased insensible losses (e.g. a febrile patient), and insufficient fluid replacement--this is the most common cause of hypotension!
Medications--especially calcium channel blockers, beta-blockers, alpha-antagonists, and diuretics.
Decreased cardiac output, as in cardiogenic shock, MI, or heart failure.
Decreased systemic vascular resistance, as in sepsis or severe metabolic acidosis.
Anemia
Hemodynamic redistribution, as in orthostatic hypotension, anaphylaxis, or neurogenic shock.
Lightheadedness and dizziness are cardinal signs of hypotension though other presenting symptoms may include:
Cool, clammy skin
Diaphoresis
Blurry vision
If the hypotension is sufficiently severe, syncope and seizures may occur
Evaluation of the hypotensive patient should begin with a repeat blood pressure measurement in both upper extremities, followed by a search for the underlying cause if the hypotension is validated.
A deviation of as little as 20 mm Hg below an individual’s baseline blood pressure can result in lightheadedness and syncope.
Hypotension of unknown etiology should be evaluated with:
Electrocardiogram (ECG)
Complete blood count (CBC)
Basic metabolic profile (BMP)
Serum lactate
The following steps should be taken in the resuscitation of a hypotensive patient:
Fluid challenge
Pharmacologic pressure support (e.g. norepinephrine, epinephrine, phenylephrine, vasopressin, etc.)
Pharmacologic inotropic support (e.g. dopamine, dobutamine, milrinone, etc.).
Consider transfusing packed red blood cells (PRBCs) for a Hgb < 7 g.
Mechanical blood pressure support systems such as an intra-aortic balloon pump (IABP)
Atherosclerosis
Atherosclerosis is the chronic inflammatory response to lipid accumulation in the intimal layer of arterial walls which over time leads to progressive hardening of the artery, loss of elasticity, and plaque formation.
The pathogenesis of atherosclerosis occurs in 3 steps:
Lipid-laden macrophages (foam cells) accumulate in the intimal layer at sites of minor vascular trauma beginning early in life, leading to formation of the fatty streak.
Plaque progression occurs as smooth muscle cells migrate into the intima in response to cytokines, de-differentiate into fibroblasts, and proliferate.
Plaque disruption may occur if the overlying endothelial layer is damaged, resulting in thrombus formation with the potential for embolization.
Atherosclerosis is the most common cause of myocardial infarctions (90%) and strokes (60%), leading to most cases of heart failure and approximately one third of dementia diagnoses.
Important risk factors for atherosclerosis include:
Family history of coronary artery disease
Cigarette smoking
High LDL
Low HDL
Type 2 diabetes mellitus
Hypertension
Obesity
It is important to recognize modifiable risk factors for the Step 2 CK.
Cigarette smoking is the most important modifiable risk factor for atherosclerosis-related heart disease.
Homocysteinemia is strongly associated with an increased rate of atherosclerotic events and disease progression. This is most commonly due to either vitamin B12 (cobalamin) or folate deficiency, but may also occur in the setting of congenital homocystinuria. In the former case, look for additional signs and symptoms of B12 or folate deficiency (neuropathy, multilobular polymorphonuclear cells, megaloblastic anemia, etc.); in the latter case, look for a patient with a marfanoid body habitus and cardiovascular disease at a young age. While controversial, some studies (and at least one USMLE World question) indicate that vitamin B6 (pyridoxine) deficiency can contribute to clinically significant alteration in homocysteine metabolism.
Large and medium-sized muscular arteries are the primary sites of atherosclerosis.
The most commonly affected vessels, in decreasing order of frequency of involvement, are:
Abdominal aorta
Coronary arteries
Popliteal artery
Internal carotid artery
Circle of Willis
Clinical presentation is dependent on the severity of ischemia, the degree of luminal occlusion, and the location of involvement.
Involvement of coronary arteries leads to myocardial infarction.
Internal carotid involvement causes transient ischemic attack (TIA) and stroke.
Atherosclerosis of the renal arteries is an important cause of secondary hypertension.
Peripheral artery involvement causes vascular insufficiency, intermittent claudication, and gangrene.
Visceral atherosclerosis leads to mesenteric ischemia.
The diagnosis of atherosclerosis is most often made by clinical manifestations of ischemia in conjunction with angiography demonstrating luminal narrowing.
Treatment of atherosclerosis is primarily oriented to managing modifiable risk factors.
The initial therapy for a new patient with risk factors and/or evidence of atherosclerotic disease is always lifestyle interventions such as smoking cessation, reducing dietary cholesterol, blood glucose and pressure control, and weight loss.
Patients without multiple risk factors who fail management with lifestyle modification after 3-6 months should begin statin therapy. Some patients with significant risk factors may be best treated with pharmacotherapy initially--see Hypercholesterolemia.
Dyslipidemia
Hypercholesterolemia is defined as an excess of low-density lipoproteins (LDL) (>130 mg/dL) or insufficient high-density lipoproteins (HDL) (<40 mg/dL).
See Lipid Digestion and Metabolism and Familial Hyperlipidemias for a review of lipid metabolism its derangements.
The LDL-C level reported in the lipid profile is an estimation based on the formula:
Hypercholesterolemia is multifactorial with a significant genetic component; however, additional factors which contribute to an elevated LDL-C level and lower HDL-C level include the following:
Obesity
Diabetes mellitus
Tobacco use
Alcohol abuse
Diets high in fatty foods
Increased estrogen exposure (including oral contraceptive pills)
Certain medications (see below)
Medications which can contribute to an elevated LDL-C level include:
Thiazide diuretics
Cyclosporine
Glucocorticoids
Amiodarone
Secondary causes of elevated LDL-C include 'REHAAB':
Renal disorders: nephrotic syndrome, uremia
Endocrine disorders: hypothyroidism, diabetes mellitus, Cushing's syndrome
Hepatocellular carcinoma
Anorexia nervosa
Acute intermittent porphyria
Biliary stasis
Several secondary causes of reduced HDL-C include malnutrition, Gaucher’s disease, and drugs (anabolic steroids, beta blockers).
Most dyslipidemia is diagnosed in an otherwise asymptomatic patient as part of routine screening.
Classic physical signs of dyslipidemia include:
Xanthelasmas: lipid deposits around the eyelids, seen in familial hypercholesterolemia (high LDL-C)
Xanthomas: lipid deposits on the trunk (eruptive--seen in familial hypertriglyceridemia), within extensor tendons (tendinous--seen familial hypercholesterolemia), and on extensor surfaces (tuberous--seen with elevated LDL-C or triglycerides)
Retinal cholesterol emboli
Corneal arcus
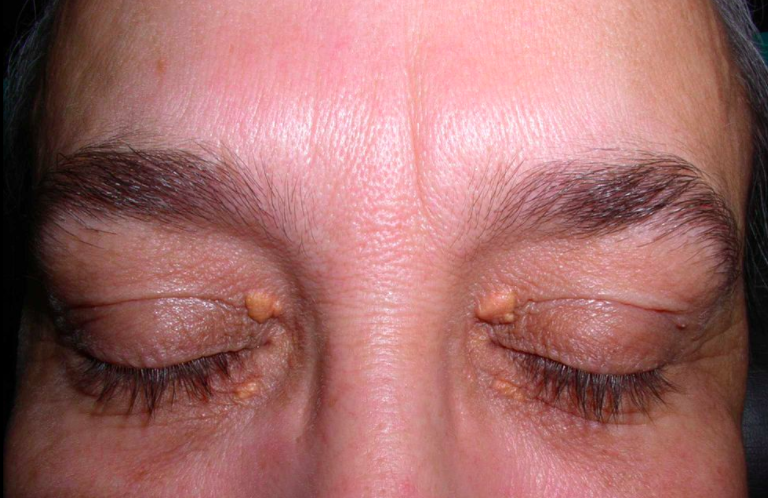
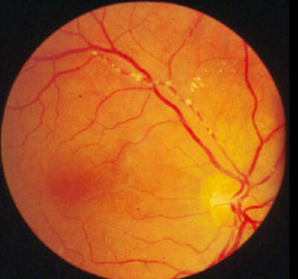
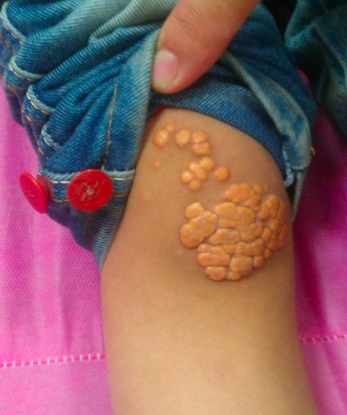
Patients with extremely elevated triglyceride levels are at risk for pancreatitis.
Current American College of Cardiology/American Heart Association (ACC/AHA) task force guidelines recommend screening for dyslipidemia with lipid profiles every five years beginning at 35 years of age in low-risk men and 45 years of age in low-risk women.
There is currently no consensus guideline regarding the age at which regular lipid testing should be implemented in higher-risk individuals; however, frequently stated ages are 20-25 years of age for high-risk men and 20-35 years of age for high-risk women.
Additionally, initial screening is appropriate in certain pediatric patients. Please refer to the Adolescent Health Screening topic card under Pediatrics.
Healthy lifestyle changes including exercise programs, weight reduction, decreasing dietary fat intake, and smoking cessationshould be recommended to _all_patients, and are considered to be the foundation of atherosclerotic cardiovascular disease (ASCVD) risk-reduction.
Pharmacologic intervention with a statin is recommended for four groups of patients:
Primary prevention of ASCVD in patients 21 years of age and older with LDL-C ≥ 190.
Primary prevention of ASCVD in patients at least 45 years of age and less than 75 years of age with diabetes mellitus and an LDL-C of at least 70.
Primary prevention of ASCVD in patients at least 45 years of age and less than 75 years of age without diabetes mellitus or history of clinical ASCVD and with LDL-C between 70 and 189 who have a ten-year ASCVD risk greater than or equal to 7.5%.
Secondary prevention in patients with clinical ASCVD (previous myocardial infarction, coronary artery disease, cerebrovascular attack, or peripheral arterial disease).
The initiation of statin therapy _in addition to lifestyle modifications_is the best initial approach to management for these patients.
The current ACC/AHA guidelines state that the use of non-statin agents did not provide any significant reduction in ASCVD risk once LDL-C goals had been achieved; however, the use of non-statin agents may be appropriate in patients with an unsatisfactory response to statins, in patients who cannot tolerate statin therapy or for whom statins are contraindicated, and/or patients with a genetic dyslipidemia syndrome. Several of these non-statin agents are reviewed in the accompanying chart. Note that these agents do commonly appear in test questions.
Cardiac Arrythmia

SVT
Supraventricular tachycardia (SVT) refers to any tachyarrhythmia that arises from a pacemaker above the Bundle of His and is characterized as a narrow QRS complex on electrocardiogram (ECG).
Paroxysmal SVT is a term used to describe SVTs characterized by an abrupt onset and termination which are generally the result of reentry circuits, though the actual mechanisms are diverse.
SVTs include many different tachyarrhthmias:
Sinus tachycardia
Atrial tachycardia
Atrial flutter
Atrial fibrillation
Multifocal atrial tachycardia (MAT)
Atrioventricular nodal reentrant tachycardia (AVNRT)
Atrioventricular reentrant/reciprocating tachycardia (AVRT)
Junctional tachycardia
Sinus tachycardia is rhythm originating in the sinoatrial node which exhibits 1:1 atrioventricular (AV) conduction (P before every QRS, QRS following every P) with a regular rate of >100 beats per minute.
Physiologic sinus tachycardia occurs in normal subjects with an increase in sympathetic tone (exercise, emotions, pain), alcohol use, caffeine ingestion, and certain drugs such as those with anticholinergic or beta-adrenergic agonist effects.
Possible pathologic causes of sinus tachycardia include:
Fever secondary to systemic illness
Hypotension, hypovolemia, or shock
Anemia
Thyrotoxicosis
Heart failure or myocardial infarction (MI)
Pulmonary embolism (Note that the most common ECG finding in the setting of pulmonary embolism is indeed sinus tachycardia--a frequently tested and pimped point.)
The diagnosis is made on review of an ECG demonstrating a sinus rhythm of greater than 100 BPM.
Treatment is directed at underlying causes (e.g. antipyretics if the cause is fever). Beta-blockers or calcium channel blockers may be considered for overly symptomatic patients.
Follow the links below to review several examples of sinus tachycardia.
Sinus tachycardia - example 1
Sinus tachycardia - example 2 - Note the saw-tooth appearance of the wave-form in leads II and V3--do not allow yourself to mistake this for 2:1 atrial flutter. Several tips to distinguish sinus tachycardia (as in this example) vs. 2:1 atrial flutter (as, for example, in this tracing) are to look for regular distortion of the PR and ST segments and negatively deflected flutter waves in the inferior leads. This webpage is dedicated to the diagnosis of 2:1 flutter. It may be more than necessary for Step 2 CK purposes, but great if you wind up in the CCU during clerkships.
Most patients with symptomatic SVT complain of palpitations, dizziness, chest pain, diaphoresis, and/or shortness of breath.
Some patients with SVT may experience hemodynamic instability with insufficient forward cardiac output, requiring emergent direct currect electrical cardioversion.
Electrophysiological testing is the gold standard and can help identify bypass tracts and aberrant conduction systems, but is often not necessary since electrocardiogram (ECG) is usually sufficient to diagnose SVT and PSVT.
General electrocardiographic features of SVT include ventricular rate > 100 bpm with narrow QRS complexes (<120 ms).
Laboratory tests including electrolyte levels, CBC, TSH, and digoxin levels are helpful in ruling out contributing conditions.
The best initial treatment of SVT in stable patients is carotid massage or vagal maneuvers (e.g. breath holding, Valsalva, urination), as these help slow AV nodal conduction by increasing parasympathetic tone.
When vagal maneuvers do not help, the next step in therapy is adenosine. Adenosine directly blocks the AV node to help interrupt the reentrant circuit. Verapamil can also be used for this purpose.
Direct current (DC) cardioversion is the preferred treatment of SVT in patients who are hemodynamically unstable (hypotension, pulmonary edema, and altered mental status).
VFIB
Ventricular fibrillation is the most serious cardiac arrhythmia, characterized by rapid, uncoordinated quivering contractions of the ventricles.
Ventricular fibrillation is most commonly associated with coronary artery disease. Other common risk factors include myocardial infarction, decreased left ventricular ejection fraction, electrolyte disturbance, long QT syndrome, and atrial fibrillation.
Ventricular fibrillation is the most common cause of mortality in patients suffering from an acute myocardial infarction (AMI).
Patients with ventricular fibrillation classically present with a sudden loss of consciousness or a comatose state. Some patients may present with symptoms (chest pain, diaphoresis) of a myocardial infarction prior to loss of consciousness.
Electrocardiogram (ECG) of patients with ventricular fibrillation reveals chaotic waveforms without the presence of P waves, QRS complexes, or T waves.
Concurrent serum laboratory tests (e.g. cardiac enzymes, potassium, calcium, magnesium, TSH, BNP, and toxicology) are extremely important to help determine the etiology.
Ventricular fibrillation results in insufficient forward cardiac output with hemodynamic collapse, leading quickly to ischemic central nervous system damage, myocardial injury, and death.
Advanced cardiac life support (ACLS) should be implemented in the setting of ventricular fibrillation: ventricular fibrillation is a shockable rhythm and a defibrillating shock should be delivered as soon as possible, followed by chest compressions for two minutes while peripheral access is established.
After two rounds of defibrillation without restoration of a stable rhythm, epinephrine (1 mg bolus) should be administered followed by another shock. 1 mg boluses of epinephrine should be delivered every 3 to 5 minutes thereafter.
In patients refractory to three rounds of defibrillation plus epinephrine, amiodarone (300 mg IV bolus) can be delivered in anticipation of a fourth shock, as this may lower the defibrillation threshold.
Note that asystole and pulseless electrical activity (electromechanical dissociation) are NOT shockable arrhythmias. All patients in cardiopulmonary arrest should have a defibrillator attached, but if either of these conditions is present then the best approach is immediate high-quality chest compressions.
Nonsustained ventricular tachycardia is a run of 3 or more consecutive premature ventricular contractions (PVCs) which spontaneously resolves in 30 seconds or less, and thus generally does not require treatment.
VTACH
Nonsustained ventricular tachycardia is typically the result of intraventricular re-entry circuits which arise when ischemic conditions alter the electrophysiological properties of the ventricular myocardium. Additional causes can include:
Infarct scarring
Acute myocardial infarction
Cardiomyopathies
Myocarditis
Drugs (cocaine)
Electrolyte disturbances
Sustained ventricular tachycardia consists of ectopic ventricular beats which occur consecutively for more than 30 seconds. Sustained ventricular tachycardia is a medical emergency, requiring immediate treatment!
The most common cause of emergent ventricular tachycardia is coronary artery disease, and it is the most common cause of death due to myocardial infarction (MI).
The clinical presentation of ventricular tachycardia ranges from asymptomatic (usually chronic) to hemodynamic compromise requiring immediate electrical cardioversion.
The characteristic appearance of ventricular tachycardia on ECG is wide, regular QRS tachycardia with QRS complexes lasting longer than 140 msec.
Note that additional wide-complex tachycardias (supraventricular tachycardia (SVT) with aberrancy, atrial tachyarrhythmia in Wolff-Parkinson-White syndrome, SVT with preexisting bundle branch block (BBB), etc.) may mimic VT, and are sometimes distinguished on the basis of the response to antiarrhythmic medication; however, all patients with hemodynamic compromise due to wide-complex tachycardia require immediate direct-current cardioversion regardless of the etiology.
The Brugada Criteria sets parameters to diagnose VT. If ANY of these criteria are met, then the rhythm is ventricular tachycardia:
No RS complex in any precordial leads
An R-to-S interval >100 ms in any precordial lead
Atrioventricular dissociation (p waves are regular but not related to the rate of QRS complexes)
Leads V1, V2 and V6 fulfilling classic criteria for ventricular tachycardia.
If NONE of these criteria are met, the rhythm is likely a wide-complex supraventricular tachycardia.
If hemodynamic compromise is present, immediate high-quality chest compressions and direct-current electrical cardioversion are indicated.
See ACLS: Cardiac Arrest for more information.
If the patient is hemodynamically stable, elective cardioversion with lidocaine, class IA antiarrhythmics, or amiodarone may be indicated for the treatment of sustained ventricular tachycardia.
AFIB
Atrial fibrillation refers to the quivering state of the atria that occurs when many ectopic atrial foci fire in a chaotic manner that prevents the normal coordinated atrial contraction.
Atrial fibrillation is the most common cardiac arrhythmia.
The single most important risk factor for atrial fibrillation is mitral stenosis.
Atrial fibrillation may be the presenting symptom of hyperthyroidism.
The atrioventricular (AV) node only conducts some of the atrial impulses through to the ventricles, resulting in the classic irregularly irregular rhythm. Though atrial rate may exceed 500 bpm, the ventricular rate is typically 120-180 bpm.
Ectopic foci around the pulmonic veins are most often implicated in the generation of atrial fibrillation.
Atrial fibrillation leads to a rapid and irregular heartbeat, which in turn causes palpitations and exercise intolerance. Since the “quivering” atria are unable to pump blood effectively, venous stasis may lead to congestive symptoms such as shortness of breath and edema.
An EKG reveals an irregularly irregular rhythm with a disorganized baseline electrical activity, the absence of p-waves, and narrow QRS complexes (since the signal originates above the AV node).
Because the blood pressure varies from beat to beat, digital blood pressure machines often have trouble getting accurate measurements in patients with atrial fibrillation.
Laboratory workup should include, at a minimum:
Testing for renal function
Electrolytes
TSH
CBC
PT/INR
Complications of atrial fibrillation arise from reduced cardiac output (loss of the atrial kick greatly reduces preload), increased cardiac oxygen demand, and thromboembolism secondary to atrial stasis.
Because the atria quiver instead of contracting, stagnant blood in the atrium may clot, leading to embolic events such as a stroke. This is most likely to occur in a region called the left atrial appendage. (Because patients with intermittent atrial fibrillation may experience return of the atrial kick, they may be more likely to embolize clots that form).
Myocardial infarction may arise secondary to increased cardiac oxygen demand.
Reduced cardiac output may lead to symptoms of congestive heart failure, such as pulmonary or lower extremity edema.
Cardioversion to sinus rhythm is indicated for the treatment of atrial fibrillation in the following settings:
For the treatment of symptomatic atrial fibrillation which continues despite the ventricular rate having been controlled with a beta-blocker or a calcium channel blocker.
For first-time occurrence of atrial fibrillation, but only if the onset is known to be within the past 2 days or if the patient has been anticoagulated for at least 3 weeks. (Patients must also be anticoagulated for 4 weeks after cardioversion).
In the setting of hemodynamic instability.
Note that the second point is extremely important and frequently tested with the 'next best step in management' type of question.
Chemical (pharmacologic) cardioversion from atrial fibrillation to sinus rhythm may be attempted chemically via class IC (propafenone, flecainide), or class III (ibutilide, dofetilide > amiodarone, sotalol) antiarrhythmic drugs.
Electrical cardioversion to sinus rhythm from atrial fibrillation may be attempted by the delivery of direct current (DC) voltage synchronized with the QRS complex.
Management strategies for atrial fibrillation may include rate control and/or rhythm control. Anticoagulation is added to prevent stroke and thromboembolism.
Warfarin is the medication of choice to prevent stroke in atrial fibrillation and the only medication approved by the FDA for the treatment of valve-related atrial fibrillation, but newer anticoagulants such as dabigatran and apixaban may be used in patients with non-valvular atrial fibrillation. Recall that anticoagulants are not the same as antiplatelet medications such as aspirin and clopidogrel (Plavix).
Coagulation studies (aPTT/INR) should be ordered before beginning anticoagulant therapy. INR is used in clinical practice to monitor warfarin levels; however, warfarin will cause a prolongation of both PT/INR as well as aPTT.
Ventricular rate control is achieved using beta blockers (class II antiarrhythmics) or calcium channel blockers (diltiazem, verapamil--class IV antiarrhythmics), which slow conduction from the fibrillating atria to the ventricles. This strategy is generally employed in patients older than 65.
Rhythm control is achieved using antiarrhythmic agents such as amiodarone, propafenone, dofetilide, flecainide, or sotalol, which alter the cardiac action potential in such a way that helps maintain sinus rhythm.
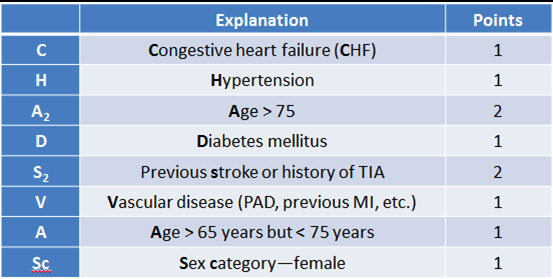
Several calculations are available to estimate the risk of stroke in patients with atrial fibrillation. These include the CHADS, CHADS2, and CHA2DS2-VASc. The CHA2DS2-VASc is the most current, and is summarized in the included table. Generally, anticoagulation is indicated for a score of 2 or greater. Aspirin monotherapy has not been demonstrated to offer significant protection against ischemic stroke for patients with low risk (CHA2DS2-VASc score < 2).
[Gregory et al. Chest. 2013;137:263-72.] [Lane, Lip. Circulation. 2012;126:860-5.]
AFLUT
Atrial Flutter is a type of supraventricular tachycardia which arises due to a macro-reentry circuit within the atrium (most commonly involving irritable foci in the right atrium near the tricuspid annulus) and is characterized by an atrial rate 250-350 BPM (classically 300 BPM).
Since the atrioventricular (AV) node has a slower conduction rate than the atrial muscle, only some of the atrial depolarizations are conducted to the ventricles, resulting in a functional AV block.The ratio of atrial to ventricular depolarizations is used to describe the rhythm (i.e. atrial rate of 340 bpm and a ventricular rate of 170 bpm would be described as a 2:1 flutter).
Atrial flutter usually occurs in the following settings:
Coronary artery disease (CAD)
Thyrotoxicosis
Mitral valve disease
Cardiac surgery
Chronic obstructive pulmonary disease (COPD)
Pulmonary embolism
Pericarditis
Generally anything that causes atrial fibrillation can cause atrial flutter.
The classic presentation of atrial flutter consists of palpitations with a rapid heart rate. While atrial flutter is often asymptomatic in otherwise healthy patients, patients with underlying heart disease may develop symptoms related to decreased cardiac output early in the disease.
On physical exam, patients with atrial flutter will have a rapid but mostly regular heart rate with a pulse usually in the range of 120-180 bpm.
Atrial flutter is recognized on electrocardiogram (ECG) by regular, sawtooth flutter waves of atrial contraction at a rate of 250-350 bpm with a ventricular response rate of approximately 180 bpm. These flutter waves are most easily seen in the inferior leads, as well as V2.
Follow the links below to review several examples of atrial flutter.
Atrial flutter - example 1 (2:1 conduction) Refer to the above discussion under sinus tachycardia examples regarding distinguishing sinus tachycardia vs. 2:1 flutter.
Atrial flutter - example 2 (3:1 conduction)
Atrial flutter - example 3 (4:1 conduction)
Persistent, untreated atrial flutter often degenerates into atrial fibrillation.
The additional strain on the heart associated with atrial flutter predisposes the patient to the same pathophysiology as atrial fibrillation:
Cardiac ischemia
Dizziness and/or syncope, secondary to diminished forward output
Decompensated heart failure, particularly in patients with preexisting cardiac disease
As in atrial fibrillation, atrial flutter that creates ineffective contraction of the atria can promote thrombus formation and predispose to thromboembolic events.
Medical management of persistent atrial flutter is the same as atrial fibrillation and includes anticoagulation and rate and/or rhythm control with direct current electrical cardioversion for patients who are hemodynamically unstable.
Atrial flutter often recurs, so ablation is generally recommended. This procedure involves mapping the re-entrant circuit that is causing the flutter rhythm, then creating a line of scar tissue that disrupts conduction through the circuit. Recall that this is usually tissue in the vicinity of the tricuspid annulus.
Long QT
Pathogenesis
Etiologies of acquired long QT syndrome include:
Drugs
Electrolyte imbalance
Myocardial infarction
Remember the mnemonic for drugs that can cause Torsades de pointes: ABCDE
AntiArrhythmics (class IA - especially quinidine & class III)
AntiBiotics (e.g. macrolides)
Anti"C"ychotics (e.g. haloperidol)
AntiDepressants (e.g. TCAs)
AntiEmetics (e.g. ondansetron)
Additionally, methadone, cisapride and arsenic trioxide can cause Torsades de pointes.
Drugs that are known to cause acquired long QT syndrome have the effect of blocking the cardiac $I_K$ current mediated potassium channel of the heart, that is, the rectifying potassium current which is responsible for the repolarization to resting membrane equilibrium potential. Low serum potassium can make this effect worse.
Specific electrolyte imbalances known to cause acquired long QT syndrome include:
Hypokalemia
Hypomagnesemia
Hypocalcemia
Note: low serum potassium can enhance a drug's inhibition of the cardiac IKr current.
Prolonged QT is diagnosed by ECG. QTc is the corrected QT interval, which is defined as the QT interval divided by the square root of the RR interval: $QTc=QT/\sqrt{RR}$ ; however, QTc is more easily estimated by checking that the QT is less than half of the RR. The normal QTc value is less than 440 milliseconds.
Torsades de pointes (TdP), a polymorphic ventricular tachycardia, is a life threatening complication of QT prolongation.
Management
The management of acquired long QT syndrome depends on the specific cause, which can include:
Stopping the offending drug if drug induced
Correcting any electrolyte abnormalities (hypokalemia, hypomagnesemia, hypocalcemia)
IV magnesium sulfate if the patient develops TdP
AVNRT
Atrioventricular Nodal Reentry Tachycardia (AVNRT) is a type of supraventricular tachycardia (SVT) which originates within the AV node.
The basis for AVNRT is the existence of two separate conduction pathways within the AV node: a slow pathway with a short refractory period and a fast pathway with a long refractory period which share a final common pathway.
In sinus rhythm, the sinoatrial impulse arrives at the AV node and conducts down both pathways simultaneously. Usually by the time the impulse traverses the slow pathway, the fast pathway is still refractory, so the impulse is extinguished.
In the common form of AVNRT, a perfectly-timed premature atrial beat arrives while the fast pathway is refractory and the slow pathway is able to conduct, thus allowing a re-entry circuit to develop if the fast pathway has recovered by the time the impulse traverses the slow pathway:
The impulse conducts down the slow pathway _but not the fast pathway, which is refractory.
The fast pathway recovers while the impulse traverses the slow pathway.
The impulse arrives at the final common pathway and proceeds in a retrograde fashion up the fast pathway, which results in retrograde atrial depolarization.
The slow pathway recovers and re-conducts the impulse to the ventricles.
AVNRT occurs mostly in young patients with healthy hearts.
AVNRT presents with the same symptoms as most SVTs:
Sudden onset tachycardia (sensation of heart racing)
Palpitations
Dizziness
Dyspnea
Chest discomfort
Presyncope/syncope
Electrocardiogram (ECG) will show a narrow-complex tachycardia (unless pre-existing BBB or aberrant ventricular conduction exists). Retrograde P-waves are present, but are often buried in or fused to the QRS complex.
ECG in between episodes of AVNRT will be the normal baseline ECG for that patient--that is, there is NO ventricular pre-excitation or characteristic ECG abnormality as there is in patients with Wolff-Parkinson-White syndrome.
Management
The long-term management of AVNRT involves, in the following order:
Rate control with diltiazem, verapamil, or a beta-blocker;
Rhythm control with antiarrhythmic drugs such as flecainide or propafenone (Class IC); and
Radiocatheter ablation for symptomatic patients failing previously-mentioned treatments.
Acute symptomatic AVNRT is best managed in the same manner as other symptomatic SVTs: if initial vagal maneuvers and carotid massage fail to restore normal sinus rhythm, administration of IV adenosine or verapamil is indicated. If at any point the patient develops signs of hemodynamic instability, direct current (DC) cardioversion should be performed immediately.
Multifocal Atrial Tachycardia
Multifocal atrial tachycardia (MAT) is an irregularly-irregular supraventricular tachycardia (SVT) characterized by the presence of 3 or more ectopic atrial pacemaking foci.
MAT occurs more commonly in patients with COPD, hypoxemia, and underlying pulmonary dysfunction. Other Less common etiological factors include hypokalemia, hypomagnesemia, sepsis, and theophylline or digitalis toxicity.
Most patients found to have MAT present with symptoms of an underlying pulmonary condition (e.g. shortness of breath, productive cough, chest pain). Only rarely do patients present with primary complaints of palpitations or syncopal episodes.
The best diagnostic test is an electrocardiogram (ECG), which will show an (irregularly) irregular rhythm with an atrial rate greater than 100 bpm and at least 3 morphologically distinct P waves on the same lead.
An ECG of MAT has the following defining features:
An atrial rate of 100-200 BPM
At least three morphologically distinct P waves occurring on the same lead
P waves return to baseline
P-P intervals, P-R duration, and R-R intervals vary
Some P waves may not be conducted, but every QRS complex is preceded by a P wave
MAT can be suspected in patients with an irregular, rapid pulse and a history of pulmonary disease.
Treatment of MAT should be directed first at the patient's underlying condition: electrolyte abnormalities should be addressed and potentially offending medications removed.
Symptomatic MAT is best managed with rate control via verapamil or cardio-selective beta blockers (e.g. metoprolol, esmolol), which allows the heart to pump more efficiently and allows for better oxygenation.
There is no role for electrical cardioversion or rhythm control with antiarrhythmics.
For patients with ongoing, symptomatic MAT who cannot tolerate pharmacologic rate control, radiocatheter ablation of the AV node and concomitant pacemaker placement provide a final means of relief.
WPW
In Wolff-Parkinson-White (WPW) syndrome a separate accessory pathway known as the Bundle of Kentis present between the atria and ventricles which allows aberrant conduction between the atria and ventricles to bypass the AV node.
The accessory Bundle of Kent conducts atrial impulses to the level of the ventriclesfasterthan the AV node, which naturally delays AV conduction.
WPW syndrome is the basis for atrioventricular(AV) reentry/reciprocating tachycardia (AVRT), a type of supraventricular tachycardia (SVT).
In orthodromic AVRT, the most common type of AVRT, the reentry circuit involves forward conduction of atrial impulses to the ventricles via the AV node with retrograde conduction from the ventricles back up to the atria via the accessory pathway.
In antidromic AVRT, the less common type of AVRT, the reentry circuit involves forward conduction down the accessory pathway with retrograde conduction up the His-Purkinje system through the AV node.
The result of the different AV conduction velocities in patients with an accessory Kent bundle is ventricular preexcitation, which is seen as a delta wave (see image) on electrocardiogram (ECG) of a patient with an accessory Kent bundle in sinus rhythm.
Symptomatic AVRT presents in the same manner as most SVTs:
Sudden onset tachycardia (sensation of heart racing)
Palpitations
Dizziness
Dyspnea
Chest discomfort
Presyncope/syncope
Orthodromic AVRT produces a regular, narrow-complex tachycardia with a ventricular rate of 150-250 BPM(may be greater) and inverted P waves on ECG. Sometimes there is a beat-to-beat oscillation in the QRS amplitude, a finding known as electrical QRS alternans, which is more commonly associated with pericardial effusion.
Antidromic AVRT produces a regular, wide-complex tachycardiawith a ventricular rate of 150-250 BPM with inverted P waves on ECG, which may be confused with ventricular tachycardia.
The management of symptomatic orthodromic AVRT in hemodynamically stable patients is first vagal maneuvers. If unsuccessful, then AV-nodal blocking agents (IV adenosine preferred over IV verapamil), and immediate direct current (DC) cardioversion for hemodynamically unstable patients.
AV nodal blocking agents such as adenosine, beta blockers, calcium channel blockers (especially verapamil), and digoxin should not be used for AF in patients with WPW as they may promote conduction across the accessory pathway and lead to degeneration of AF into VF.
The management of symptomatic antidromic AVRT in hemodynamically stable patients is with IV procainamide.
Definitive management of AVRT is radiocatheter ablation of the accessory Bundle of Kent.
Bradycardia
Sinus bradycardia is a regular sinus rhythm characterized by a resting heart rate of < 60 BPM.
Physiologic sinus bradycardia can be caused by sleep, athletic conditioning, and normal vagal activity.
Pathological sinus bradycardia may be caused by:
Exaggerated vagal tone
Sinoatrial (SA) node ischemia (e.g. myocardial infarction in the right coronary artery (RCA) distribution)
Sick sinus syndrome
Increased intracranial pressure (ICP)
Hypothyroidism
Hypothermia
Medications (e.g. calcium channel blockers, beta-blockers)
Sinus bradycardia is usually asymptomatic, especially in the conditioned athlete; however, progressive pathological bradycardia may result in fatigue, presyncope, or syncope.
Treatment is NOT indicated in asymptomatic patients with sinus bradycardia. Management of symptomatic patients involves removing offending agents and treating underlying causes when these are present. Pacemakers should be considered for patients with intrinsic SA node damage.
Sick Sinus
Sick Sinus Syndrome (SSS) is the condition characterized by chronic SA node dysfunction (marked bradycardia, sinus pause/arrest, sinoatrial block) secondary to senescence of the node and surrounding myocardium.
SSS is most commonly associated with SA node ischemia, such as might occur secondary to a proximal right coronary artery (RCA) infarction (see Coronary Anatomy). Other causes include inflammatory processes or infiltrative diseases that involve the SA node.
Sick sinus syndrome generally presents in older patients who have multiple comorbidities and high mortality rates with:
Chronic and often severe bradycardia
Sinus pauses, sinus arrest, and SA nodal exit block
Tachy-brady syndrome--alternating bouts of bradycardia and atrial tachyarrhythmias (most commonly atrial fibrillation)
Patients may complain of lightheadedness, syncope, exertional dyspnea, and worsening angina.
The diagnosis is often clinical, with the characteristic symptoms and accompanying signs being the major clue. An ECG is always obtained but is nonspecific. Look for marked sinus bradycardia (p waves present) with occasional dropped beats (absent p-qrs-t), supraventricular escape rhythms, atrial fibrillation (i.e. the tachy-brady syndrome), and evidence of localized ischemia (especially in the RCA territory).
Symptomatic bradycardia in the setting of sick sinus syndrome is an indication for pacemaker placement. Pharmacological treatment of tachyarrhythmias with beta-blockers, calcium channel blockers, and/or digoxin should be considered; however, these medications may exacerbate sinus bradycardia. Note that while lightheadedness and syncope are reversed in almost all patients following pacemaker placement, there does not seem to be a survival benefit.
[Birnie D, Williams K, Guo A, et al. Reasons for escalating pacemaker implants. Am J Cardiol 2006;98:93.]
Junctional Rhythm
Junctional rhythm or AV nodal rhythm is a pathological bradycardic rhythm that occurs when the SA node fails to fire at a normal rate. It is often called a “junctional escape rhythm” because the AV node “escapes” from the pacing of the SA node, which normally fires with a faster intrinsic rate to serve as the primary pacemaker of the heart and override lower pacemakers.
Junctional escape rhythms most commonly arise in the setting of profound SA node dysfunction, when the degree of sinus bradycardia is below the intrinsic rate of the SA node. These bradyarrhythmias occur at a characteristic rate of 40-60 BPM, which is the intrinsic rate of the AV node. (Less commonly, in the setting of abnormally increased junctional automaticity, junctional tachycardia may out-pace a normal sinus rate. This can occur in the setting of digitalis toxicity, recent cardiac surgery, acute myocardial infarction, or isoproterenol infusion.)
Patients will often be asymptomatic; however, lightheadedness and syncope can occur as a consequence of decreased ventricular filling and cardiac output secondary to uncoordinated contraction of the atria with the ventricles, and should prompt an electrocardiogram (ECG). The hallmark of junctional rhythms on ECG is the absence of P waves preceding the QRS complex. P waves may be incorporated in with or occur after the QRS complex, indicating retrograde depolarization of the SA node.
Treatment

The decision of whether or not to place a pacemaker for the treatment of bradycardia is guided by two main factors: (1) association of symptoms with arrhythmia and (2) the potential for progression of arrhythmia disturbance.
Class I indications for pacemaker placement in the management of sinus bradycardia include:
Symptomatic sinus bradycardia
Sinus bradycardia which prevents exercise (symptomatic chronotropic incompetence)
Symptomatic Mobitz I or II
Mobitz type II with widened QRS or chronic bifascicular block, regardless of symptoms
Advanced second degree AV block (block of two or more consecutive P waves)
Complete (third degree) AV block
Exercise-induced second or third degree AV block
Class II indications for pacemaker placement in the management of sinus bradycardia include:
First degree AV block with hemodynamic compromise
Asymptomatic Mobitz Type II AV block with a narrow complex QRS
Persistent, asymptomatic second- or third-degree AV block following myocardial infarction
Heart Failure
RHF
Right heart failure (RHF) can be thought of as "backwards" failure, meaning the right ventricle cannot pump blood into the lungs, causing blood to accumulate in the systemic venous system. The most common cause of right heart failure is left heart failure. Other causes include: pulmonary hypertension, left-to-right shunts, and tricuspid valve regurgitation.
The most common cause of right heart failure in the absence of left heart failure is COPD.
Right heart failure caused by chronically elevated pulmonary arterial pressures (often related to interstitial lung disease) is known as cor pulmonale.
Right sided heart failure is generally said to be a disease of signs (rather than symptoms like in LHF.) The signs are caused by venous congestion and include hepatosplenomegaly, peripheral edema, and jugular venous distension.
Hepatomegaly is due to venous congestion of hepatic veins of the liver. If the venous congestion is severe enough, it can cause portal hypertension, which can lead to splenomegaly and ascites.
The presence of hepatojugular reflux, in which pressing on the RUQ of the abdomen elicits distention of the right jugular vein, indicates right heart failure, strongly suggesting a cardiac cause of hepatomegaly (rather than an intrahepatic cause, such as cirrhosis).
Peripheral edema is caused by an increase in venous hydrostatic pressure, resulting in pitting edema of dependent areas (e.g. lower limbs).
Jugular venous distention is caused by increased venous pressure in the superior vena cava, resulting in increased visible distension of the jugular veins. This is generally measured with the head of the bed at 45º, in centimeters from the sternal notch. >4cm is considered abnormal.
The above findings can also be seen in left heart failure that has also progressed into right failure.
Diagnosing right heart failure begins with left heart failure investigations, as that is the most common cause. Diagnosis of cor pulmonale (right heart failure secondary to pulmonary arterial hypertension) is discussed below.
The ECG in severe pulmonary hypertension shows P pulmonale (peaked P waves > 2.5 mm in the inferior leads II, III and AVF), right axis deviation, and right ventricular hypertrophy.
Chest x-ray will show right ventricular enlargement and dilatation of the main pulmonary artery, hilar vessels, and descending right pulmonary artery. There will be no vascular congestion or pulmonary edema, which would be consistent with left heart failure (the most common cause of right heart failure, but NOT cor pulmonale!).
Echocardiography with Doppler is a good first screening tool to evaluate right venticular chamber size and wall thickness, and may demonstrate right ventricular hypertrophy with paradoxical displacement of the interventricular septum into the left ventricle during systole, and eventually tricuspid regurgitation with progressive dilatation of the right ventricle.
BNP will be elevated in acute right heart failure, as in left heart failure.
Treatment for acute right heart failure is the same as left heart failure, and they often coexist. Treatment for cor pulmonale can be remembered by "Oh, Acid Is Work":
Adequate Oxygenation (oxygen saturation >90-92%)
Correct respiratory Acidosis (will dilate pulmonary vasculature and reduce right heart afterload)
Treat underlying Infections
Decrease work of breathing by using non-invasive positive pressure ventilation and/or bronchodilators
Heart Blocks
Atrioventricular (AV) blocks occur when there is a delay or interruption in the normal conduction of an electrical impulse from the atria to the ventricles due to anatomical or functional impairment of the AV node.
First degree atrioventricular (AV) block occurs when the PR interval is greater than 0.2 seconds with a normal (1:1) P:QRS ratio. Note that this is the size of one large box on standard electrocardiogram (ECG) paper.
First degree AV block is usually the result of increased vagal tone (most common) or functional conduction impairment (e.g. AV nodal disease, electrolyte disturbances, or medication side effects).
First degree AV block is typically asymptomatic, and is discovered on ECG obtained for another purpose.
It is diagnosed strictly by an ECG showing a PR interval greater than 0.2 seconds, which equals one big box or five small boxes on ECG paper.
Generally no treatment is necessary, but investigations into secondary causes like electrolyte disturbances or medications may be warranted.
However, a PR interval >300 msec can have hemodynamic consequences,and these patients may benefit from pacing.1 Additionally, note that the presence of a widened QRS complex is indicative of pathology below the AV node, and these patients will require further evaluation.2
1[Epstein et al. Circulation. 2008;117.] 2[Sauer. UpToDate. Updated 31 Oct 2014.]
Second-Degree AV block occurs when some atrial impulses are not conducted to the ventricles. The two subtypes are Mobitz Type I (Wenkebach) and Mobitz Type II.
Mobitz I results from an intranodal or HIS bundle conduction defect that results from medications (e.g. beta blockers, digoxin, calcium channel blockers), increased vagal tone, or right coronary artery mediated ischemia. Mobitz II results from an infranodal conduction abnormalityin either the bundle of His or Purkinje fibers.
Both Mobitz I and II are typically asymptomatic, though patients may experience a "skipped beat".
Mobitz I (Wenkebach) block displays progressive PR lengthening until a QRS complex is dropped. Group beating may also be observed--this refers to the clumping of P-QRS-T elements leading up to the dropped QRS complex. Mobitz II displays a seemingly random dropped QRS (i.e. there is NO progressive PR lengthening). There is generally a discernible ratio of P:QRS (e.g. two P waves for every one QRS, or 2:1), but not always.
Mobitz I block is treated by adjusting medications or pacing if associated with symptomatic bradycardia. Mobitz II block is always treated with a pacemaker due to the increased risk of progressing to a high grade or third degree (complete) AV block.
Third degree (complete) heart block (i.e. AV dissociation) occurs when supraventricular impulses completely fail to conduct to the ventricles, and ventricular depolarization is initiated by pacemaker cells distal to the block. It is represented by P waves that are wholly independent from the QRS complex.
Complete heart block typically results from coronary ischemia, resulting in progressive degeneration of the conducting system; however, it may result from congenital AV block, lupus, or Lyme disease.
Third degree AV block presents with syncope, dizziness, and hypotension.
The ECG will show no relationship between P waves and QRS complexes. Note that PP and RR intervals are constant; however, PR intervals vary.
These patients require a pacemaker and should also avoid drugs that further affect the AV node and its conduction.
Dilated Cardiomyopathy
Dilated cardiomyopathy occurs when the left or both ventricle(s) become(s) massively dilated with a disproportionate loss of cardiac muscle mass, leading to decreased contractility and systolic dysfunction.
Dilated cardiomyopathy is the most common cardiomyopathy, accounting for as many as 90% of all cardiomyopathies.
Other causes of dilated cardiomyopathy include (mnemonic: ABCCCCDDE):
Alcohol abuse
wet Beriberi
Chagas disease
Cocaine
Coxsackie B virus
peripartum Cardiomyopathy
Doxorubicin, Daunorubicin
hEmochromatosis
The most commonly mutated gene in dilated cardiomyopathy is TTN, which encodes the protein, titin.
The most common mutation in X-linked dilated cardiomyopathy is dystrophin.
The presentation of dilated cardiomyopathy is similar to that of congestive heart failure (CHF):
Orthopnea
Dyspnea
Weakness
Edema
Weight gain
Dilated cardiomyopathy classically causes a S3 heart sound.
Patients will often have regurgitant murmurs, which result from the stretched annuli as the heart dilates.
Electrocardiogram (ECG) of patients with dilated cardiomyopathy may demonstrate first degree atrioventricular (AV) block, left bundle branch block (LBBB), left anterior fascicular block, or a nonspecific intraventricular conduction abnormality.
Chest X-ray will show a massive balloon-like heart.
Dilated cardiomyopathy is managed symptomatically with diuretics, ACEIs, and β-blockers, though the definitive treatment is a heart transplant.
Carditis and Cardiomyopathy
Infective Endocarditis
Infective endocarditis is the infection of the endocardial surface of the heart which typically involves the cusps of valves.
Acute infective endocarditis is most commonly caused by S. aureus and occurs on normal heart valves. If left untreated, it is rapidly fatal in <6 weeks.
Subacut infective endocarditis is caused by less virulent organisms like viridans group streptococci, Staph. epidermidis, enterococci, and organisms of the HACEK group of bacteria infecting damaged or abnormal valves. Even if left untreated, subacute endocarditis is typically not rapidly fatal.
The HACEK group of bacteria consists of the following organisms:
Haemophilus spp.
Actinobacillus
Cardiobacterium
Eikenella
Kingella
These organisms are difficult to culture; thus, endocarditis caused by these organisms is sometimes referred to as "culture negative".
Nonbacterial thrombotic endocarditis is a group of non-infectious endocarditides in which sterile vegetations of platelets and thrombi form on valves. These are friable and highly susceptible to embolization.
Nonbacterial thrombotic endocarditis is most commonly caused by malignancy (marantic endocarditis), followed by systemic lupus erythematosus (Libman-Sacks endocarditis).
Libman-Sacks endocarditis is form of nonbacterial thrombotic endocarditis in which sterile verrucous lesions of thrombi and immune complexes form on both surfaces of the valve in patients with systemic lupus erythematosus.
These lesions can cause systemic emboli and regurgitant murmurs (e.g. mitral, aortic regurgitation).
Risk factors for infective endocarditis include prosthetic heart valves, damaged or abnormal valves, and IV drug use.
Endocarditis in IV drug users is more commonly right-sided, and may be caused by cutaneous flora and fungi in addition to the more common Staph. aureus.
Strep. bovis includes 4 species of group D strep that classically cause endocarditis in patients with colonic lesions, including cancer. Of these, S. gallolyticus has the strongest association between colonic lesions and endocarditis.
Organisms of the viridans group of Streptococcus spp. together with enterococci are the most common cause of subacute infective endocarditis on native valves. The viridans group includes the following organisms:
Streptococcus mitis
Streptococcus oralis
Streptococcus sanguinis
Streptococcus sobrinas
Streptococcus milleri
Note that some questions will offer one of these species instead of "viridans group streptococci" as an answer choice.
Surgical instrumentation and manipulation of the genitourinary tract can be associated with enterococcus endocarditis. (Recall that enterococci are among the two most common causes of infectious endocarditis on native valves, with the other being viridans group streptococci.)
The diagnosis of infective endocarditis is made using the Duke criteria:
Major criteria:
Sustained bacteremia
Evidence of endocardial damage: vegetation on echocardiography, new regurgitant murmur
Minor criteria:
Predisposing conditions: abnormal valves, predisposition to bacteremia, history of IV drug abuse
Fever
Vascular phenomena: septic emboli, mycotic aneurysms, intracranial hemorrhage, Janeway lesions, splinter hemorrhages
Immune phenomena: glomerulonephritis, positive rheumatoid factor, Osler nodes, Roth spots
Use the mnemonic bacteria FROM JANE to help recall these signs and symptoms of infective endocarditis:
Fever (most common symptom)
Roth spots (retinal hemorrhages with pale centers)
Osler nodes (tender raised lesions on toes and fingers)
Murmur (mostly Mitral)
Janeway lesions (nontender erythematous lesions on sole and palm)
Anemia (usually anemia of chronic disease)
Nailbed hemorrhage
Emboli (infarcts in different tissues)
When infective endocarditis is suspected, at least two sets of blood cultures should be drawn. It has been shown that three sets are superior to two, but the yield is not significantly increased with four.
Immunologic manifestations of infective endocarditis include:
Glomerulonephritis (hematuria, red cell casts)
Osler nodes (tender raised lesions on toes and fingers)
Roth spots (retinal hemorrhages with pale centers)
Rheumatoid factor
Until cultures and sensitivity data can confirm the appropriate choice of antibiotics, empiric therapy is instituted with vancomycin.
Appropriate initial antibiotic therapy for culture-proven viridans group streptococci and Strep. bovis on native and prosthetic valves consists of one of the following:
Penicillin G + gentamicin
Ceftriaxone + gentamicin
Vancomycin for patients with penicillin allergy
Note that gentamicin is optional for native valve endocarditis if the strain is penicillin-susceptible.
Appropriate initial antibiotic therapy for culture-proven Staph. spp. endocarditis consists of one the following:
Oxacillin + gentamicin
Nafcillin + gentamicin
Cefazolin + gentamicin
Vancomycin for patients with penicillin allergy or MRSA endocarditis.
Note that gentamicin is optional for native valve endocarditis; however, if the patient has a prosthetic valve, then gentamicin AND rifampin are indicatedin addition to the treatment described above.
According to the 2009 ACC guidelines, antibiotic prophylaxis for endocarditis should ONLY be given for:
Congenital cyanotic lesions which have not been surgically corrected
Prior valve repair using prosthetic material
Prior history of endocarditis
History of heart transplant
According to the 2009 ACC guidelines, patients with known ventricular septal defects, atrial defects, and mitral valve prolapse DO NOT require antibiotic prophylaxis prior to undergoing invasive procedures.
In addition to treating the underlying disorder, nonbacterial thrombotic endocarditis is treated with anticoagulation with heparin.
Myocarditis
Myocarditis is caused by any inflammatory process involving the cardiac muscle. Usually this is an upper respiratory virus.
Viral infections are the most common cause of myocarditis. These include:
Coxsackie B virus _(_most common)
Parvovirus B-19 (also important to remember)
Echovirus (sometimes 'enterovirus' more generically)
Adenovirus
EBV
CMV
HHV-6
Though more rare (and not commonly tested), additional infectious etiologies of myocarditis include:
Bacterial: Diphtheria (toxin-related), Klebsiella spp. Salmonella spp. Brucella spp. Legionella pneumophila, streptococci (scarlet fever), staphylococci, pneumococci, tuberculosis, Borrelia burgorferi
Rickettsial
Fungal (coccidiodomycosis, histoplasmosis, candidiasis)
Protozoal (esp. Trypanosoma cruzi)
It can also be caused by toxicity of the following drugs:
Doxorubicin
Cyclophosphamide
Chloroquine
Penicillins
Sulfonamide
Cocaine
Myocarditis in a patient with ties to South and Central America should prompt a consideration of Trypanosoma cruzi infection (called Chagas disease). These patients may have additional symptoms similar to achalasia.
The presentation of myocarditis is highly variable, ranging from subclinical to heart failure and sudden cardiac death. Many patients have a recent history of upper respiratory tract infection.
A feared complication of myocarditis is dilated cardiomyopathy, which may present with sudden onset of extreme fatigue, dyspnea, and decreased exercise capacity in a previously healthy patient.
Chest pain in the setting of myocarditis is usually due to concomitant pericarditis and may mimic acute coronary syndrome in severity.
Palpitations and various arrhythmias are common in the setting of myocarditis, with atrial tachycardia being the most frequent.
Physical examination findings associated with myocarditis may include:
S3 or S4 heart sound as signs of ventricular dysfunction
If severely dilated, murmurs of tricuspid or mitral insufficiency
Pericardial friction rub indicating concomitant pericarditis
Heart failure signs and symptoms (rales, jugular venous distention, etc.) due to dilated cardiomyopathy and ventricular dysfunction.
Displacement of the point of maximal impulse (PMI), consistent with cardiomegaly.
Important diagnostic tests to order when considering a diagnosis of myocarditis include electrocardiogram (ECG), cardiac biomarkers, and echocardiogram. A chest x-ray should be obtained, but is usually normal.
Electrocardiogram (ECG) may be normal or reveal nonspecific findings in the setting of myocarditis, including ST segment and T wave abnormalitiesthat mimic myocardial infarction. Involvement of the pericardium may produce global ST segment elevation and related findings.
Elevated cardiac biomarkers (troponins) indicate the presence of myocardial necrosis, while persistance indicates ongoing disease.
Echocardiography is the most valuable diagnostic modality in the assessment of myocarditis, and may demonstrate an enlarged, spherical heart with wall motion abnormalities and systolic dysfunction.
Viral titers and serology may be helpful in determining the particular infectious agent.
Myocardial biopsy will show myocyte inflammation with monocytes and macrophages and focal areas of necrosis.
Treatment of myocarditis involves addressing the root cause(such as stopping an offending medication) and supportive carefor any associated symptoms of heart failure.
Immunosuppressive agents such as corticosteroids, azathioprine, or cyclosporine may be beneficial because of the observation that inflammation often persists following clearance of the pathogen.
Pericarditis
Pericarditis occurs when an inflammatory process involves the pericardial sac.
Pathogenesis
Serous (effusive) pericarditis occurs when underlying autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis), viral infection (e,g. pt with recent URI comes in with signs of dyspnea, fatigue, increased cardiac silhouette), trauma, or uremia causes the accumulation of transudative or exudative fluid in the pericardial sac.
Note that some serous effusions may contain red blood cells (i.e. serousanguinsous), but that this is considered distinct from hemorrhagic pericarditis.
Fibrinous pericarditis occurs when underlying uremia, recent myocardial infarction, acute rheumatic fever, or radiation injury causes accumulation of a fibrin-rich exudate within the pericardial sac.
Purulent pericarditis occurs when a bacterial infection of the pericardial space causes an accumulation of pus in the pericardial sac.
Hemorrhagic pericarditis refers to the presence of gross blood in the pericardial sac, and is generally due to underlying malignancy; however, tubercular pericarditis is a common etiology in endemic regions.
Symptoms
The typical presentation of acute pericarditis is pleuritic chest pain of sudden onset that is worsened by deep inspiration and relieved by sitting forward. Pain may radiate to the trapezius muscle. Additional signs and symptoms may point to an underlying etiology, such as a viral illness, autoimmune process, or malignancy.
PE
Characteristic findings on physical examination include a multiphasic friction rub best heard at the left lower sternal border with diaphragm of the stethoscope and distant heart sounds if a pericardial effusion is present. May have nonpalpable point of maximal impulse.
Large pericardial effusions may compress the left lower lobe of the lung, resulting in atelectasis. This is appreciated as 'woody' dullness to percussion, egophony, and auscultation of bronchial breath sounds at the left inferior scapular angle, which in this setting is known as Ewart's sign.
Diagnosis
A diagnosis of acute pericarditis is generally suspected on the basis of supportive history & physical exam findings, but electrocardiogram (ECG) is the best additional test to confirm the diagnosis.
The characteristic ECG in the setting of acute pericarditis will show:
Global ST segment elevation (sensitive)
PR depression (specific)
Diffuse low voltage and electrical alternans, a beat-to-beat variation in QRS amplitude which indicates the presence of a pericardial effusion
Note that some test questions will try to trick you into diagnosing a massive myocardial infarction--look for a friction rub or other clues which point more to pericarditis.
Chest radiograph should always be obtained in the setting of suspected pericarditis. It is usually normal, but acute pericarditis should always be suspected when the chest X-ray reveals otherwise unexplained cardiomegaly in the appropriate clinical setting. Water bottle heart with clear lung fields:

Though a normal echocardiogram does not rule out pericarditis, it is always necessary to:
Determine whether tamponade physiology is present
Characterize purulent pericarditis (i.e. presence of loculations)
Distinguish pericardial stranding from fibrin or tumor, and myocarditis from pericarditis
Additional initial laboratory evaluations include complete blood count (CBC), cardiac biomarkers, and inflammatory markers. These are discussed in the accompanying chart.
Treatment
The first-line treatment for most patients with acute idiopathic or viral pericarditis is colchicine plus NSAIDs (e.g. ibuprofen). In patients with acute pericarditis following a myocardial infarction, aspirin is preferred to other NSAIDs.
Glucocorticoids are used in patients with pericarditis that is refractory to NSAIDs plus colchicine as well as in patients with autoimmune pericarditis, pericarditis secondary to connective tissue disease, and uremic pericarditis that is unresponsive to dialysis.
Purulent pericarditis requires administration of intravenous antibiotics as soon as it is recognized, generally followed by drainage via subxiphoid pericardial windowing or pericardiectomy.
Chronic Constrictive Pericarditis
Chronic constrictive pericarditis occurs when a chronic inflammatory process involving the pericardial space results in consolidation and scarring.
The pathophysiologic effect of chronic constrictive pericarditis is principally on diastole. Ventricular systolic function is essentially normal.
The clinical picture of chronic constrictive pericarditis develops insidiously with evolving fibrinous organization of the pericardium. Gradually patients display symptoms of diminishing cardiac output and elevated systemic venous pressures:
Diminishing cardiac output leads to fatigue, hypotension, and reflex tachycardia.
Elevated systemic venous pressures lead to jugular venous distention (JVD) ,hepatomegaly with ascites, and peripheral edema.
Constrictive pericarditis can be distinguished from hepatic cirrhosis by the presence of jugular venous distention (JVD) on physical examination in patients with constrictive pericarditis, as well as by measurement of the protein content of the ascitic fluid via paracentesis: hepatic ascites has a low protein content (< 2.5 g/dL), whereas cardiac ascites will has a higher protein content (> 2.5 g/dL).
Physical examination may reveal jugular venous distention (JVD) and paradoxical increase in JVD with inspiration (Kussmaul’s sign). Cardiac auscultation may reveal an early diastolic knock and a friction rub. Note that pulsus paradoxus is more classically associated with cardiac tamponade, and does not occur as commonly in the setting of constrictive pericarditis.
The constricted ventricles do not accommodate the physiologic increase in venous return which normally occurs with inspiration. Instead of entering the right ventricle, the increased inspiratory venous return ‘backs up’ through the right atrium into the systemic veins. This causes the physiologically paradoxical_finding of neck vein distention with _inspiration_instead of expiration which is known as _Kussmaul’s sign.
Constrictive pericarditis is suggested by a thickened pericardium (> 2 mm) on chest CT. The diagnosis is confirmed by cardiac catheterization.
Calcification of the pericardial space may be appreciated on chest CT. This radiographic finding is accompanied by an early diastolic knock on cardiac auscultation.
Cardiac catheterization reveals rapid early diastolic filling of the ventricles which quickly plateaus, as well as a prominent y descent on the right atrial pressure tracing. In contrast, cardiac tamponade characteristically demonstrates a blunted y descent on the right atrial pressure tracing.
In untreated chronic constrictive pericarditis, the pericardial layers eventually fuse, resulting in an inelastic pericardium which cannot accommodate normal filling of the ventricles.
The constricted pericardium reduces right ventricular end diastolic volume (RVEDV), leading to a rise in systemic pressures and signs of right heart failure.
Once fibrinous organization of the pericardial space has occurred, the only treatment is surgical pericardectomy.
Though currently rare in developed countries, tuberculous constrictive pericarditis should not be overlooked, especially in immunocompromised individuals and patients from regions in which Mycobacterium tuberculosis is endemic. These patients require quadruple drug therapy.
Constrictive pericarditis can produce similar clinical and hemodynamic findings to restrictive cardiomyopathy. Don't be fooled! Please use the chart in the images section to help you distinguish between these two entities.
Restrictive Cardiomyopathy
Restrictive cardiomyopathy refers to a dysfunctional state in which a stiff, noncompliant ventricle with normal wall thickness results in severe diastolic dysfunction, restrictive filling with elevated filling pressures, and largely preserved systolic function.
Infiltrative disorders that may cause restrictive cardiomyopathy include:
Amyloidosis
Sarcoidosis
Gaucher disease
Hurler syndrome
Non-infiltrative disorders that may cause restrictive cardiomyopathy include:
Storage diseases (hemochromatosis, Fabry disease, glycogen storage diseases)
Diabetic cardiomyopathy
Scleroderma
Carcinoid heart disease
Endomyocardial fibrosis (hypereosinophilic syndrome)
Restrictive cardiomyopathy will present with characteristic heart failure symptoms, but may be confused with constrictive pericarditis. Many patients (especially those presenting on NBME examinations) will have associated conditions like amyloidosis, sarcoidosis, hemochromatosis, diabetes, etc.
Restrictive cardiomyopathy is diagnosed through a combination of history, echocardiography, cardiac catheterization, and right ventricular endomyocardial biopsy. Additional imaging may include cardiac MRI, PET, and CT, depending on the clinical situation.
Physical exam of a patient with restrictive cardiomyopathy is often indistinguishable from that of a patient with constrictive pericarditis. Both patients have an elevated jugular venous pressure with a prominent y descent, and both may have a positive Kussmaul's sign. The left ventricular impulse is normally palpable over the precordium with restrictive cardiomyopathy, but will generally be deadened or nonpalpable in the setting of constrictive pericarditis.
Echocardiography of a patient with restrictive cardiomyopathy will usually show normal thickness myocardium(but maybe thickened in certain infiltrative processes) with impaired diastolic filling; ejection fraction is often preserved.
Cardiac MRI, PET, and CT are useful in the diagnostic evaluation of restrictive cardiomyopathy to identify granulomas, inflammation, and edema. CT may additionally detect pericardial calcifications, a distinguishing feature of constrictive pericarditis.
Cardiac catheterization of a patient with restrictive cardiomyopathy will show increased ventricular filling pressures with a classic dip-and-plateau pattern in the pressure tracing.
The two primary complications associated with restrictive cardiomyopathy are heart failure and arrhythmia.
First-line treatment of restrictive cardiomyopathy is directed at controlling the contributing pathology, so it is important to determine whether or not such is present.
Patients with syncopeor ventricular arrhythmias may warrant an internal cardiac defibrillator.
Digoxin should be avoided in patients with restrictive cardiomyopathy secondary to cardiac amyloidosis because it is bound by extracellular amyloid and may result in digoxin hypersensitivity or toxicity.
Cardiac sarcoidosis is treated with glucocorticoids, though this does not increase survival.
The definitive treatment for restrictive cardiomyopathy is heart transplant.
HCM
Hypertrophic cardiomyopathy (HCM) is a genetic disorder of the myocardiumcaused by mutations in the genes responsible for coding elements of the contractile apparatus which results in asymmetric, disorganized thickening of the myocardium in the absence of an identifiable cause. Note that HCM can occur both with and without outflow tract obstruction, and terms such as "hypertrophic obstructive cardiomyopathy (HOCM)" are no longer in use.
Familial HCM is inherited in an autosomal dominant pattern.
The most common pathogenic allele implicated in HCM is a mutation of the cardiac myosin binding protein C gene, which is associated with variable penetrance with some individuals remaining asymptomatic and others presenting in adulthood. The second most common pathogenic allele is a mutation in the cardiac beta-myosin heavy chain gene, which is associated with higher penetrance, younger age of onset, and more severe hypertrophy.
There is an extremely high incidence of HCM in patients with Friedreich’s ataxia.
HCM is the most commonly inherited cardiovascular disease.
The presentation of HCM ranges from asymptomatic to sudden cardiac death.
A young athlete with no previous cardiac history who suddenly collapses and is pulseless on the playing fieldshould be considered to have HCM until proven otherwise, particularly on examinations.
Approximately ⅓ of patients will report a positive family history for HCM, though due to the heterogeneity of the disease the true genetic incidence is likely much higher.
The auscultatory findings in patients with HCM vary based on the degree of left ventricular outflow tract obstruction. When obstruction is present, a medium-pitched systolic crescendo-decrescendo ejection murmur is auscultated along the left lower sternal border and apex. Patients without a subaortic gradient may not have a murmur at all.
When present, the murmur is intensified with physical activity and Valsalva, and reduced by rest, squatting, and lying down.
The variability of the outflow obstruction (and thus the murmur) in relation to exercise as well as a characteristic lack of radiation to the carotid arteries can be useful in distinguishing the murmur of HCM from that of aortic stenosis.
The diagnosis of HCM is made with echocardiography, which demonstrates a asymmetric hypertrophy of the interventricular septum and a variable degree of dynamic left ventricular outflow tract obstruction.
Arrhythmiasand sudden cardiac deathare the two most severe complications associated with hypertrophic cardiomyopathy, and are frequently the presenting symptom.
Hypertrophic cardiomyopathy is treated symptomaticallywith the goal of reducing sudden cardiac death.
Asymptomatic individuals are typically not treated.
Surgery or alcohol ablation relieve symptoms by reducing the outflow obstruction.
Beta-blockers and non-dihydropyridine calcium channel blockers (e.g. diltiazem and verapamil) are the primary pharmacological therapies, and can be used prophylactically in athletes.
Patients should avoid strenuous exercise and drugs that increase the left ventricular outflow gradient (e.g. dihydropyridine calcium channel blockers, diuretics, nitrates, and vasodilators).
Takayasu Arteritis
Takayasu arteritis is a chronic, primarily cell-mediated vasculitis affecting the aorta and its primary branches.
The initial lesion of Takayasu arteritis occurs most commonly in the proximal ⅔ of the left subclavian artery, but can progress to involve any or all of the aorta and essentially all of its primary branches.
Ongoing inflammation can cause progressive vascular thickening, leading to stricture and stenosis, as well as destruction of the elastic lamina and muscular media, leading to aneurysmal dilatation.
The majority of patients affected by Takayasu arteritis are women(80%-90%), and the disease is more common in those of Asian ancestry.
Patients with Takayasu arteritis appear chronically ill upon presentation with a variety of non-specific, non-cardiovascular complaints early in the natural history, including the following:
_C_onstitutional symptoms of fatigue, weight loss and low-grade fever;
_A_rticular manifestations resembling juvenile idiopathic arthritis;
_D_ermatologic lesions resembling erythema nodosum and pyoderma gangrenosum.
Vascular symptoms of Takayasu arteritis do not manifest until later in the natural history! Eventually vascular insufficiency results in CANCAMS:
_C_ool extremities
_A_rm and leg claudication
_N_eurovascular compromise (vertigo, syncope, orthostasis, headaches, seizures, dementia, and eventually visual impairment)
_C_oronary artery disease (CAD) and acute coronary syndromes (ACS)
_A_ortic regurgitation
_M_esenteric ischemia
_S_ubclavian steal
The subclavian steal refers to retrograde vertebral artery blood flow ipsilateral to a stenotic or occluded subclavian artery that serves as an alternative supply to the ipsilateral arm, but may have deleterious neurological effects on the ipsilateral posterior circulation which are exacerbated by exercising the arm.
It may help you recall that the earlier symptoms of Takayasu arteritis are non-specific and non-vascular with the vascular symptoms occurring only later if you can remember the mnemonic CAD at start to CAD in heart, where the first CADis the collection of Constitutional, Articular, and Dermatologic symptoms described above, and the second CAD is for the usual Coronary Artery Disease (i.e. ‘in heart’).
The diagnosis of Takayasu arteritis is predominantly clinical, and is made with 90.5% sensitivity and 97.8% specificity when a patient presents with at least three of the following six criteria:
Age younger than 40 years at onset of disease
Claudication of the extremities
Diminished pulsation of one or both brachial arteries
Difference of at least 10 mm Hg in systolic pressure between the two arms
Presence of subclavian or abdominal bruit
Narrowing of the aorta or proximal arteries on arteriography which cannot be attributed to another etiology
Note that a significant difference in systolic pressure between the two upper extremities is not specific for Takayasu arteritis--other entities which may demonstrate this finding include aortic coarctation and aortic dissection.
Cardiac auscultation of the patient with Takayasu arteritis may reveal the diastolic murmur of aortic regurgitation.
Consistent (but non-specific) laboratory findings include elevated inflammatory markers (erythrocyte sedimentation rate, C-reactive protein) in the presence of a normal white count.
The initial therapy for patients with Takayasu arteritis is glucocorticoids, but only about 50% of patients typically respond.
For Takayasu arteritis refractory to glucocorticoid therapy, consider the addition of methotrexate, azathioprine, or other such disease-modifying antirheumatic drugs (DMARDs).
Angioplastyor bypass graftingshould be considered for high-risk irreversible arterial stenoses.
Heart Murmurs
Mitral Stenosis
Mitral stenosis is narrowing of the mitral valve due to calcification or valvular damage that results in an increased pressure gradient between the left atrium and left ventricle.
Mitral stenosis results in increased left atrial pressures which are eventually transmitted to the pulmonary venous circuit.
Almost all cases of mitral stenosis are secondary to rheumatic heart disease, an autoimmune condition which results in scarring and narrowing of the valve with eventual fusion of the commissures. This is sometimes called a “fish mouth” valve.
Symptoms and PE
Backup behind the stenotic mitral valve eventually involves the pulmonary venous vasculature, leading to exertional dyspnea, orthopnea, and paroxysmal nocturnal dyspnea.
Hemoptysis due to mitral stenosis can occur if the increased pulmonary circuit pressures rupture small pulmonary vessels, spilling blood into the alveoli.
Symptoms are exacerbated by increased cardiac demand.
On auscultation a low-pitched diastolic rumble following an opening snap just after the second heart sound is heard best at the fourth intercostal space, midclavicular line, with the patient in left lateral decubitus position. The murmur is often followed by a loud S1.
The time between the opening snap and the second heart sound decreases as the stenosis becomes more severe. Patients may have a loud first heart sound by comparison.
Diagnosis
Chest X-ray will demonstrate an enlarged left atrium with straightening of the left heart border, pulmonary congestion and elevation of the mainstem bronchus due to mass effect of the enlarged atrium. Lateral chest x-ray projection can reveal posterior displacement and impingement of the esophagus.

Echocardiography is necessary to confirm and stage disease
Pressure gradient
Valve area
Valve score (0-16) based on leaflet mobility and thickening, subvalvular thickening, and calcification
Mitral stenosis may cause right-sided heart failure or stroke.
Thromboembolic stroke secondary to mitral stenosis may occur if the dilated atrium causes atrial fibrillation, leading to stagnant blood that forms a mural clot (often in the left atrial appendage).
Right sided heart failure can result from mitral stenosis when the pressure backup behind the stenotic valve extends into the pulmonary circuit and causes the pulmonary vasculature to constrict, leading to pulmonary hypertension.
Treatment
Initial management of mitral stenosis involves the use of medications to reduce intravascular volume with the goal of reducing pulmonary vascular congestion.
Loop diuretics and salt restriction are used to relieve symptoms of pulmonary congestion, and β-blockers may help relieve dyspnea. Anticoagulation with warfarin is indicated for patients with atrial fibrillation, atrial thrombus, or a previous embolic event.
Surgical management is indicated for severe disease:
Heart failure symptoms with mitral valve area (MVA) <1.5cm2
Heart failure symptoms with MVA >1.5 but increased PASP/PCWP
Change in pressure gradient across MV with exercise
Asymptomatic patients with MVA<1.5 and pulmonary HTN
Options for surgery include percutaneous balloon valvuloplasty (balloon open the valve) or (when contraindicated) open commissurotomy (open surgery) and mitral valve replacement.
Mitral stenosis is no longer considered a high risk heart condition which requires antibiotic prophylaxis for things like dental procedures. An exception to this would be if a patient with mitral stenosis has a history of infective endocarditis.
Mitral Regurgitation
Pathogenesis
Mitral regurgitation, also known as mitral insufficiency, refers to the retrograde flow of blood through the mitral valve from the left ventricle to the left atrium during systole due to anomalies of the mitral valve or the papillary muscles.
Myocardial infarction involving supply to the papillary muscles can cause them not to function properly, leading to mitral insufficiency.
Mitral valve prolapse is the most common cause of mitral regurgitation as the prolapsed and “floppy” valve is unable to hold against the pressure differences experienced during ventricular systole.
Intrinsic valvular damage can be caused by infective endocarditis and rheumatic fever.
Diseases that cause dilation of the left ventricle (e.g. aortic stenosis, aortic regurgitation) can lead to mitral regurgitation by stretching the mitral valve annulus.
Rarely, ergotamine, pergolide, and cabergoline can cause mitral regurgitation. Note that this is far less common than other etiologies, and should only be considered as a possibility after having ruled out others definitively--even if a patient is taking one of these drugs!
Symptoms
Acute mitral regurgitation presents with jugular venous distention and sudden onset of congestive heart failure, while chronic disease presents with an apical thrill without signs of congestive heart failure.
Acute mitral regurgitation presents with the rapid onset of severe congestive heart failure with a low cardiac output, and is commonly due to rupture of a recently infarcted papillary muscle (i.e. a patient who has suffered a myocardial infarction within the past couple of days).
In acute onset mitral regurgitation, the left atrium is unable to remodel rapidly enough to accommodate the increased volume. This leads to a rapid increase in left atrial filling pressure, resulting in increased pulmonary capillary pressure and pulmonary edema.
Patients with chronic mitral regurgitation may be asymptomatic with normal exercise tolerance; however, they are often sensitive to shifts in volume status and may be at risk for development of acute volume overload (flash pulmonary edema) and right-sided heart failure.
Mitral regurgitation causes a high-pitched holosystolic murmur at the apex with radiation to the axilla. Remember that mitral regurgitation often occurs in patients with mitral valve prolapse, so the murmur of mitral regurgitation may co-occur with a mid-systolic click.
Diagnosis
Electrocardiogram (ECG) findings in chronic disease may include notched P waves referred to as P mitrale, which may also be observed in the setting of mitral stenosis and other left atrial overload states. Over time, mitral regurgitation may lead to atrial fibrillation.
Dilation of the left atrium in chronic mitral regurgitation leads to an enlarged cardiac silhouette on chest X-ray.
The diagnostic modality of choice is echocardiography with color Doppler, which demonstrates regurgitant flow.
Mitral regurgitation may cause a widely split S2 as the regurgitant flow leads to earlier emptying of the left ventricle, allowing the aortic valve to close much earlier than the pulmonic valve.
Mitral regurgitation can be associated with an S3 heart sound.
Treatment
Medical management is indicated in symptomatic patients with the goal of reducing afterload and thus regurgitant flow. Agents used include:
Diuretics
Nitrates (also reduce preload)
ACE inhibitors or ARBs
Beta blockers
Open mitral valve replacement is curative, and is indicated in the setting of very severe, symptomatic disease. Synthetic valves require ongoing anticoagulation and biosynthetic valves have a limited lifespan, making valve repair the treatment of choice in patients with a valve intact enough to withstand the procedure.
MVP
Mitral prolapse occurs when excess mitral valve leaflet tissue or myxomatous degeneration of valve components causes the mitral valve leaflets to balloon into the left atrium during systole.
Anxiety and panic disorders have been associated with mitral valve prolapse.
Myxomatous degeneration is an increase in spongiosa (composed of dermatan sulfate) and a decrease in fibrosa (which has more tensile strength) which results in redundant valvular tissue.
Connective tissue disorders such as Ehlers-Danlos, Marfan, or osteogenesis imperfecta predispose to mitral valve prolapse.
Symptoms
The majority of patients are asymptomatic, but some may complain of palpitations and chest pain.
On auscultation a mid-to-late systolic click which varies with maneuvers (earlier with decreased left ventricular end-diastolic volume (LVEDV), later with increased LVEDV) and a soft late systolic murmur of mitral regurgitation are appreciated.
Diagnosis
Echocardiography is the most useful tool in diagnosing and evaluating MVP. It can determine the degree of prolapse, severity of mitral valve thickening, presence and amount of regurgitation as well as other determinants of cardiac function such as LVEF.
Standing and valsalva decrease preload, reducing the LVEDV and relieving the chordae tendineae of some tension. This makes the click occur earlier in systole.
Squatting increases preload, augmenting the LVEDV and placing greater tension on the chordae tendineae. This makes the click occur later in systole.
Like any cardiac valvular disease, mitral valve prolapse may predispose to endocarditis, but it is very rare and the current recommendation is against antibiotic prophylaxis.
Asymptomatic patients should be reassured that their condition is benign.
Pulmonic Stenosis
Pathogenesis
Pulmonic Stenosis is a congenital disorder characterized by a narrowing of the right ventricular outflow tract or defect of the pulmonary valve that leads to decreased blood flow from the right ventricle into the pulmonary artery.
Pulmonic stenosis can occur at three locations: valvular, subvalvular, supravalvular.
In valvular pulmonic stenosis, the trileaflet valve is present with varying degrees of fibrous thickening and fusion of commissures. There is primary and secondary valvular pulmonic stenosis. Unlike the aortic valve, calcification of the pulmonary valve is rare.
Primary valvular pulmonic stenosis can be associated with congenital heart defects such as tetralogy of Fallot, congenital rubella syndrome and Noonan syndrome.
Secondary valvular pulmonic stenosis is much less common than primary (congenital) pulmonic stenosis, and can be due to carcinoid syndrome, in which case it is frequently accompanied by tricuspid regurgitation (TIPS for tricuspid insufficiency and pulmonic stenosis).
Subvalvular pulmonic stenosis results in limited blood flow through the right ventricular outflow tract due to fibromuscular hypertrophy below the pulmonary valve.
Supravalvular pulmonic stenosis results in decreased blood flow to the lungs due to areas of narrowing within the pulmonary artery or its branches.
Symptoms
Patients with pulmonic stenosis typically present with dyspnea on exertion, fatigue, and cyanosis in severe cases.
Physical examination of a patient with pulmonic stenosis may reveal the following:
Prominent jugular venous A wave
Systolic ejection murmur loudest at the upper left sternal border
Wide splitting of the second heart sound, due to increased outflow duration
Right-sided S4
In newborns, increased right ventricular pressures can cause right-to-left shunting through a patent foramen ovale or ventricular septal defect causing cyanosis.
Asymptomatic patients in childhood can present later in life with signs and symptoms of right heart failure due to untreated pulmonic stenosis.
A transesophageal echocardiogram and doppler study confirms the diagnosis of pulmonic stenosis.
A transesophageal echocardiogram will reveal the outflow obstruction, right ventricular hypertrophy, and systolic doming of the valve due to decreased movement during systole.
A doppler will allow for assessment of decreased blood flow across the obstruction.
Prolonged untreated pulmonic stenosis may result in right sided heart failure.
Right sided heart failure is due to the progressive weakening of the right ventricle caused byincreased ventricular pressures. The weakening of the right ventricle leads to fluid backup throughout the venous and portal system.
Treatment of pulmonic stenosis consists of balloon valvotomy to widen the outflow tract from the right ventricle into the pulmonary artery.
Surgery is indicated over balloon valvotomy when pulmonic stenosis is caused by a dysplastic pulmonary valve.
Neonates with severe stenosis may require a patent ductus arteriosus to maintain oxygenation. This is achieved with synthetic prostaglandin E1, such as alprostadil.
Tricuspid Regurgitation
Tricuspid regurgitation occurs when the tricuspid valve allows retrograde flow of blood from the right ventricle into the right atrium during systole.
A small, clinically insignificant amount of tricuspid regurgitation is present in about 70% of adults and can be considered a variant of normal anatomy.
Most clinically significant tricuspid regurgitation is functional; elevation of right ventricular end-systolic pressure (RVESP) causes progressive dilatation of the right atrium and ventricle thereby causing distortion of the annulus. This leads to valvular insufficiency, despite otherwise_anatomically normal_leaflets and chordae. This may be due to right ventricular pathology, pulmonary hypertension, or left heart failure.
Intrinsic valvular damage can be caused by infective endocarditis and rheumatic fever, but are a much less common cause of tricuspid regurgitation than dilation of the annulus.
Tricuspid regurgitation is also associated with Ebstein’s cardiac anomaly, a birth defect associated with lithium exposure in utero.
Tricuspid regurgitation is usually asymptomatic.
Some patients with more severe tricuspid regurgitation may present with a pulsating sensation in the neckfrom distended jugular veins and signs of right heart failure, such as peripheral edema, ascites, and painful hepatosplenomegaly.
Tricuspid regurgitation causes a holosystolic murmur heard best at the right or left mid-sternal borderthat becomes louder with maneuvers that increase venous return (e.g. inspiration). An extremely dilated right ventricle may have an associated right-sided S3.
Occasionally the systolic murmur is transmitted through the portal veins to the liver.
A pulsatile liver is virtually pathognomonic of tricuspid regurgitation. This is due to pulsatile flow through the portal vein, which may also be detected ultrasonographically.
Echocardiographyis the gold standard for diagnosing tricuspid regurgitation and may reveal valvular abnormalities, regurgitant flow, and associated right and/or left heart pathology.
Chest X-ray isusually normal, but may show an enlarged superior vena cava, right atrium, or right ventricle.
Electrocardiogram (ECG) may demonstrate nonspecific ST segment and T wave abnormalities over the precordial leads, consistent with right ventricular dysfunction.
The retrograde pressure wave from flow of blood back into the right atrium during systole may be transmitted to the jugular veins, leading to giant “v” or “cv” waves on jugular venous pulse tracing.
Prominent “cv” waves on jugular venous pulse tracing are pathognomonic of tricuspid regurgitation.
Medical therapy is the first-line management for symptomatic tricuspid regurgitation. Appropriate agents include:
Diuretics(usually loop diuretics, such as furosemide with or without an aldosterone antagonist)
ACE inhibitors/angiotensin-receptor blockers (ARBs)
β-blockers
Digitalis
Tricuspid regurgitation in the setting of infective endocarditis requires treatment with an appropriate antibiotic.
Tricuspid regurgitation secondary to pulmonary arterial hypertension (PAH) may improve with reduction of the pulmonary arterial pressure. This may be achieved through agents like phosphodiesterase inhibitors (sildenafil, tadalafil) or endothelin receptor antagonists (bosentan) in the setting of primary PAH, or mitral balloon valvuloplasty in the setting of mitral stenosis.
Valve repair or replacement surgery is indicated for tricuspid regurgitation in patients with:
Right ventricular dysfunction
Carcinoid syndrome
Ebstein anomaly
Severe/symptomatic disease refractory to medical management
Annuloplasty is indicated when dilatation of the valve annulus is the underlying cause of the regurgitation, and symptoms are severe.
Tricuspid Stenosis
In tricuspid stenosis narrowing of the tricuspid valve results in a persistent diastolic pressure gradient between the right atrium to the right ventricle.
The most common cause of acquired tricuspid stenosis is rheumatic fever.
Congenital tricuspid atresia typically presents with early cyanosis and additional associated congenital heart defects which determine the severity and phenotype.
The presentation of clinically significant tricuspid stenosis depends on whether or not there is accompanying mitral valve disease.
Most patients with clinically significant tricuspid stenosis do also have accompanying mitral valve pathology as well as some degree of tricuspid regurgitation, and present with abdominal discomfort, hepatomegaly, and hepatic congestion. Patients may also complain of a fluttering sensation in the neck caused by tall jugular venous A waves.
In the absenceof mitral valve disease, a patient with clinically significant tricuspid stenosis may present with fatigue and signs of systemic venous hypertension which are characteristically out of proportion to the dyspnea.
Tricuspid stenosis (TS) causes a diastolic rumble at the left fourth interspace. As in mitral stenosis, it may be accompanied by an opening snap.
The intensity of the murmur increases with maneuvers that increase preload and enhance the diastolic pressure gradient, including inspiration, leg raise,inhalation of amyl nitrate, and squatting.
Although Carvallo’s sign (increased murmur intensity with inspiration) is most often associated with tricuspid regurgitation, it is a feature of all right-sided heart murmurs--including tricuspid stenosis.
Chest X-ray may show dilatation of the right atrium.
Echocardiography with doppler demonstrates limited mobility of the tricuspid leaflets, diastolic doming of the valve, and high-pressure turbulent flow across the narrowed opening.
Isolated, asymptomatic tricuspid stenosis does not require treatment; however, patients with symptoms of systemic venous hypertension and congestion should be considered for balloon valvotomy.
If there is accompanying symptomatic mitral or aortic valve disease for which valvuloplasty is indicated in a patient with signs and symptoms of tricuspid stenosis, then replacement of the tricuspid valve is indicated at the time of surgery.
Aortic Regurgitation
Aortic regurgitation, also known as aortic insufficiency, refers to the retrograde flow of blood from the aorta into the left ventricle during diastole due to anomalies in the aortic valve or the aortic root.
Pathogenesis
Acute aortic regurgitation is caused by either damage to the valve leaflets or dilation of the aortic root, or both in some cases. In the developed world, the most common causes are:
Aortic root enlargement (usually trauma or aortic dissection)
Congenital bicuspid aortic valve
Calcific valve disease
In the developing world, the most common cause is rheumatic fever.
Connective tissue diseases, syphilitic aortitis, and aging can lead to chronic dilation of the aortic root, resulting in aortic regurgitation.
Symptoms
Acute aortic regurgitation presents with the rapid onset of severe congestive left heart failure, cardiovascular collapse, and manifestations of cardiogenic shock (profound hypotension, pallor, and diaphoresis) with a thready pulse and normal to reduced pulse pressure.
In chronic aortic regurgitation, the left ventricle dilates to accommodate the volume of the backflow without increasing filling pressure or left atrial pressure. These individuals may be asymptomatic until eventually the heart can no longer compensate and symptoms of congestive left heart failure develop.
Since aortic regurgitation leads to congestive left heart failure, it is exacerbated by volume overload conditions (such as a high-salt diet) and strenuous exercise.
Aortic regurgitation causes a high-pitched diastolic murmur, often with a blowing quality, beginning immediately after A2. It may decrescendo or persist throughout diastole, and is enhanced by the patient leaning forward and holding his breath at end-expiration.
PE
The spot at which the murmur is best appreciated depends on the cause of the regurgitation:
Aortic regurgitation secondary to valvular insufficiency produces the characteristic murmur best auscultated along the left sternal border at the third or fourth interspace.
Aortic regurgitation secondary to aortic root dilatation produces diastolic murmurs best auscultated along the right sternal border or at the apex.
The Austin-Flint murmur is a low-pitched mid- to late-diastolic rumble best heard at the apex that is associated with aortic regurgitation. It is caused by the turbulence of the anterograde stream from the left atrium competing with retrograde flow across the insufficient aortic valve.
The Austin-Flint murmur is best heard with the patient in the left lateral decubitus position, as this brings the left ventricle closer to the chest wall.
The Austin-Flint murmur of chronic aortic regurgitation can be distinguished from the murmur of mitral stenosis by the absence of the loud S1 and opening snap which characterize mitral stenosis.
Clinical signs commonly associated with chronic aortic regurgitation include:
Head bobbing (the force of the systolic pressure with each heartbeat causes the head to bob)
Pulsating nail bed (pulsations are visible in the nail beds due to the widened pulse pressure)
Water hammer pulse (bounding and forceful pulse that is commonly associated with widened pulse pressure)
Patients with chronic aortic regurgitation have a widened pulse pressure (high systolic blood pressure with a low diastolic blood pressure).
Diagnosis
Electrocardiogram will show left ventricular hypertrophy, left atrial dilation, and abnormal repolarization with ST segment depression at rest or during exercise.
Dilation of the left ventricle leads to an enlarged cardiac silhouette and in severe cases aortic dilation on chest x-ray.
Echocardiography shows a dilated left ventricle with regurgitation visible on color Doppler, and is the gold standard for diagnosis and staging.
Pulsus bisferiens (or biphasic pulse) refers to 2 strong systolic peaks of the aortic pulse from left ventricular ejection separated by a midsystolic dip. It can be palpated in patients with significant aortic regurgitation (with or without aortic stenosis), hypertrophic obstructive cardiomyopathy, and, occasionally, large patent ductus arteriosus.
Chronic aortic regurgitation is often associated with an S3 heart sound.
In acute onset aortic regurgitation, rapid decompensation causes flash pulmonary edema. In chronic aortic regurgitation, the left ventricular end-diastolic volume increases slowly enough that the ventricle is able to remodel, so pulmonary edema is seen only later.
Management
Medical management of aortic regurgitation is aimed at reducing afterload, but plays a limited role because symptomatic patients require valve replacement.
In acute aortic regurgitation patients generally require emergent valve replacement. Temporary stabilization may be provided with IV nitroprusside to reduce afterload and dopamine or dobutamine for inotropic support. Note that intra-aortic balloon pumps are CONTRAINDICATED because they worsen the retrograde filling of the ventricle when the balloon inflates during diastole. Left-ventricular assist devices (LVADS) provide no therapeutic benefit.
Surgical replacement of the valve is indicated for symptomatic patients prior to the development of heart failure. If the aortic root is involved it is simultaneously replaced with a composite graft.
Medical management with a low-sodium diet, ACE inhibitors, and calcium channel blockers (nifedipine) is appropriate for patients with chronic, asymptomatic aortic regurgitation and an ejection fraction > 50%.
Note that beta-blockers are relatively contraindicated due to their tendency to lengthen diastole, thereby worsening the regurgitation.
Recall that patients with midsystolic murmurs (grade ≤2) without associated findings and symptoms do not require further workup for a murmur.
In contrast, patients with murmurs with any of the following characteristics require further evaluation with echocardiography:
Holosystolic murmurs
Diastolic murmurs
Continuous murmurs
Murmurs grade ≥3
Murmurs with concomitant cardiopulmonary symptoms
Last updated