Pretest Endocrine
Sexual precocity
true sexual precocity:
Cause
girls: mature gonads respond to GnRH with sex steroids; early onset normal puberty
boys: CNS lesions
hypothalamic hamartomas: both genders, secrets GnRH
symptoms: secondary sexual characteristics. Breast, hair, odor, acne
isolated premature precocity:
cause: ovarian tumors, exogenous estrogens
symptoms: isolated premature breast and/or vaginal bleeding without pubic hair, body odor, and acne.
Cushing
external
bilateral adrenal hyperplasia
adrenal carcinoma is infant
CFTR diagnosis
The sweat test is diagnostic in virtually all cases of classic CF and is equally useful in all ethnic groups, but it is not useful in detecting heterozygotes.
DNA testing is of value for postmortem investigations as well as in sick premature infants and other patients for whom sweat collection is unsuccessful.
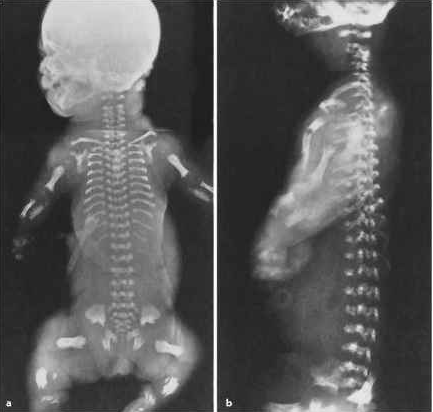
Jeune Syndrome
Aka asphyxiating thoracic dystrophy
symptoms: short stature, long and narrow thorax, hypoplastic lungs, fibrotic liver, and short limbs. Death is common, a result of pneumonia or asphyxia because of the abnormally shaped thorax.,
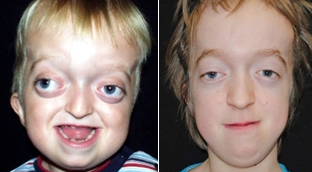
Crouzon
path: autosomal dominant
symptoms: craniosynostosis (usually coronal), proptosis, brachycephaly, hypertelorism and strabismus, “beak” nose, midface hypoplasia, and high and narrow palate.,
Achondroplasia inheritance
Achondroplasia is inherited in an autosomal dominant manner, but most cases represent spontaneous mutations in unaffected parents.
Hypercalcemia bone fracture
path: Hypercalcemia can develop in children who are immobilized following the fracture of a weight-bearing bone.
symptoms:
serious: nephropathy, nephrocalcinosis, hypertensive encephalopathy, and convulsions.
The early symptoms of hypercalcemia—namely, constipation, anorexia, occasional vomiting, polyuria, and lethargy—are nonspecific and may be ascribed to the effects of the injury and hospitalization. Therefore, careful monitoring of these patients with serial measurements of the serum ionized calcium and the urinary calcium to creatinine ratio is critical during their immobilization.
diagnosis: A ratio of greater than 0.2 Ca to Cr ratio establishes a diagnosis of hypercalciuria.
treatment:
Although complete mobilization is curative, additional measures, such as vigorous intravenous hydration with a balanced salt solution, dietary restrictions of dairy products, and administration of diuretics, can be instituted.
For patientswho are at risk for symptomatic hypercalcemia, short-term therapy with calcitonin is highly effective in reducing the concentration of serum calcium by inhibiting bone resorption.
case: A 15-year-old boy has been immobilized in a double hip spica cast for 6 weeks after having fractured his femur in a skiing accident. He has become depressed and listless during the past few days and has complained of nausea and constipation. He is found to have microscopic hematuria and a blood pressure of 150/100 mm Hg. Which of the following is the most appropriate course of action?
Gynecomastia adolescent boy
path: It is thought to be caused by a temporary reduction in the testosterone to estradiol ratio.
pt: Gynecomastia is a common occurrence in adolescent boys, especially during Tanner stage 2 or 3.
symptoms: It can occur unilaterally or bilaterally, and can affect one breast more significantly than the other.
prognosis: Spontaneous regression usually occurs; itrarely lasts for more than 2 years.
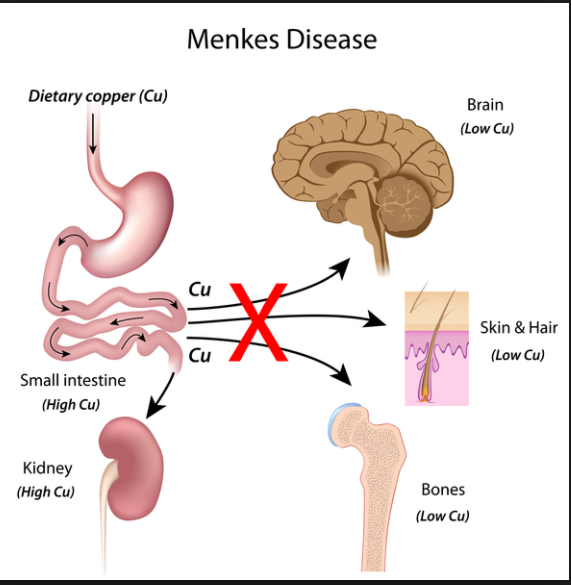
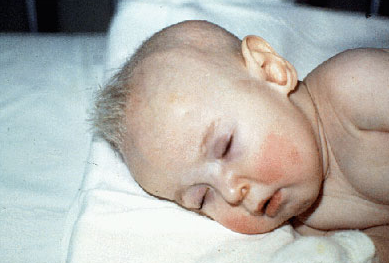
menke
path: copper absorption/transport problem being the cause
symptoms: presents in the first months of life and includes hypothermia, hypotonia, and myoclonic seizures. These children have chubby, rosy cheeks and kinky, colorless, and friable hair. Severe mental retardation is always seen.
diagnosis: Low serum copper and ceruloplasmin levels are found.,
Rickets
high PTH, hypocalciuria, hyperphosphaturia, normocalcemia, hypophosphatemia
Intestinal absorption of calcium and phosphorus is diminished in vitamin D deficiency.
Transient hypocalcemia stimulates the secretion of parathyroid hormone and the mobilization of calcium and phosphorus from bone;
enhanced parathyroid hormone activity leads to phosphaturia and diminished excretion of calcium.
In children with nutritional rickets, the concentration of serum calcium usually is normal and the phosphate level is low. Increased serum alkaline phosphatase is a common finding. The excretion of calcium in the urine is increased only after therapy with vitamin D has been instituted.
Hypocalcemia in Newborn
Hypocalcemia of newborn infants can be divided into two groups:
early (during the first approximately 72 hours of life)
late (after approximately 72 hours).
early:
The most common type of early neonatal hypocalcemia is the so-called idiopathic hypocalcemia.
Other causes early on include maternal illness (diabetes, toxemia, and hyperparathyroidism), neonatal respiratory distress (perinatal asphyxia) or sepsis, low birth weight because of prematurity, or hypomagnesemia.
late:
Transient or permanent hypoparathyroidism and high phosphate intake are the most common factors associated with late hypocalcemia.
Ca
low PO4, normal Ca
The boys in the first question have rickets unresponsive to vitamin supplementation; vitamin D–resistant rickets is caused by a genetic abnormality in the renal tubular reabsorption of phosphate with resultant hyperphosphaturia and hypophosphatemia and also in the conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. The intestinal absorption of phosphate is also abnormal, and calcium absorption from the gut can be secondarily affected. Calcium concentration is usually normal. The disorder is usually transmitted as an X-linked dominant trait.
Medullary carcinoma of thyroid arises from the C cells of the thyroid. These tumors secrete excessive amounts of calcitonin, and, accordingly, the concentration of this hormone in the blood is increased. Despite elevated levels of calcitonin, the serum concentrations of calcium and phosphorus are usually normal unless the patient has associated hyperparathyroidism (multiple endocrine adenomatosis, type II).
Vit D Def
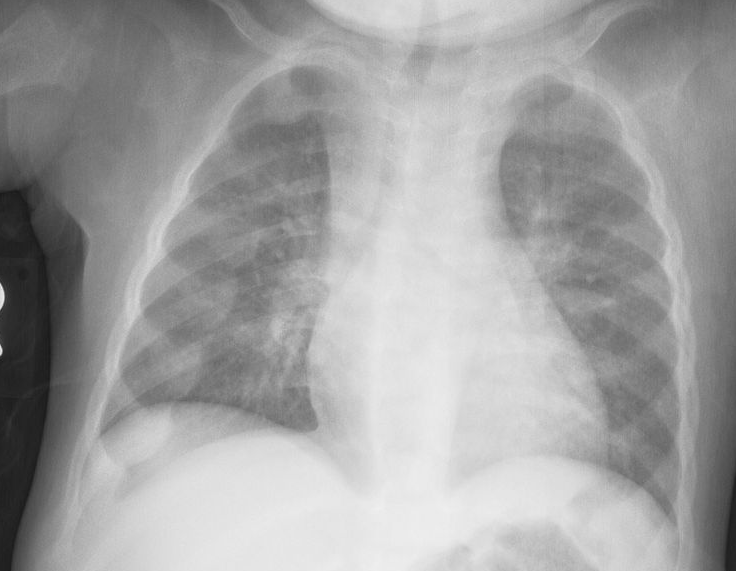
.,
Rachitic rosary: The prominent knobs of bone
Prader
path: A deletion of a portion of chromosome 15 has been found in approximately 70% of patients., The enormous food intake of affected children is thought to be caused by a defect in the satiety center in the hypothalamus.
symptoms: hypotonia in utero; poor feeding, hypogonadism, hypotonia at birth; hyperphagia after newborn
treatment: Stringent caloric restriction is the only known treatment.
Laurence-Moon-Biedl
path: autosomal recessive trait
symptoms: Obesity, mental retardation, hypogonadism, polydactyly, and retinitis pigmentosa with night blindness are the principal findings in affected children.
treatment: There is no known effective treatment.,
aka bardet biedl

Fröhlich syndrome
aka adiposogenital dystrophy
symptoms: is a rare cause of childhood obesity
association: associated with a hypothalamic tumor.,
21 Hydroxylase deficiency Na/K
In the salt-losing variety of 21-hydroxylase deficiency, the synthesis of both mineralocorticoids (eg, aldosterone) and cortisol is impaired. Aldosterone deficiency impairs the exchange of potassium for sodium in the distal renal tubule. Affected patients have hyponatremia and hyperkalemia. Dehydration, hypotension, and shock may be present.
DI Na/K
In the absence of vasopressin (central diabetes insipidus), renal collecting tubules are impermeable to water, resulting in the excretion of hypotonic urine. Patients with diabetes insipidus present with polyuria and polydipsia. Net loss of water leads to dehydration and hemoconcentration and, therefore, to relatively high serum concentrations of sodium and potassium.
Patients with nephrogenic diabetes insipidus have similar laboratory findings. This genetic disorder is unresponsive to antidiuretic hormone (ADH). These patients are unable to concentrate their urine and present in the neonatal period with hypernatremic dehydration
G6P Def Na/K
Patients with a deficiency of glucose-6-phosphatase (von Gierke disease) are, as a rule, hyperlipidemic. Increased triglyceride concentration in the serum decreases the volume of the aqueous compartment. Because electrolytes are present only in the aqueous compartment of the serum but are expressed in milliequivalents per liter of serum as a whole, the concentrations of sodium and potassium can be factitiously low in these patients.
Holt-Oram
Characterized by abnormalities in the upper extremities, hypoplastic radii, thumb abnormalities, and cardiac anomalies. Occasionally the pectoralis major muscle is missing.

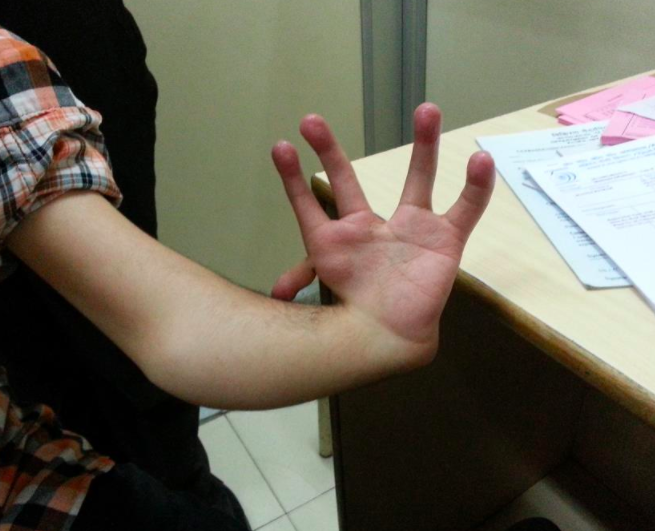
case: A newborn has low sloping shoulders, right hand attached at elbow with agenesis of the forearm, cardiac abnormalities, missing chest wall musculature, and a bifid thumb.,
Last updated