Malignancies
Metastatic
Malignancies that metastasize to the brain will occur in 10-30% of cancer patients. >50% of all brain tumors originate somewhere else in the body.
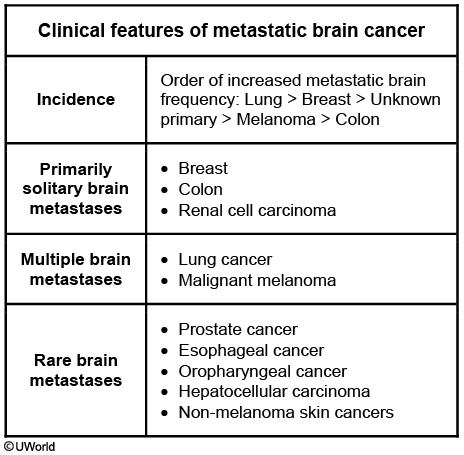
The five types of tumors that most commonly metastasize to the brain are (in order of descending frequency):
Lung (most common cause of brain metastases)
Melanoma
Renal cell cancer
Breast cancer
Colorectal cancer
The most common mechanism of metastasis to the brain is hematogenous spread. Metastases are usually located directly at the junction of the gray matter and white matter where blood vessels decrease in diameter and trap tumor cells.
Symptoms
Common presenting symptoms of metastatic brain cancer mimic those of primary CNS tumors and include:
Seizures: chronic smoker presents with grandmal seizures
Headaches
Behavioral changes
Cognitive decline
Motor or sensory deficits
Diagnosis
Diagnosis of metastatic brain cancer is made via MRI and biopsy to determine the origin of the tumor.
Radiographic features that can help differentiate brain metastases from other CNS lesions include the following:
Multiple lesions
Localization at the junction of the grey and white matter
Circumscribed margins
Primary central nervous system lymphoma is an uncommon form of non-Hodgkin lymphoma seen primarily in patients with HIV or other forms of immunocompromise. Imaging typically reveals a periventricular (not gray and white matter junction like in metastatic cancer) lesion.
Treatment
Treatment of metastatic brain cancer includes:
Treatment of the original tumor
Resection of single metastasis
Palliative radiation
For patients with a single brain metastasis in a surgically accessible location and good performance status, surgical resection offers the best therapeutic option. This is typically followed by stereotactic radiosurgery (SRS) or whole brain radiation therapy (WBRT) to the tumor bed. SRS may be used as an alternate therapeutic option for patients who are not candidates for surgery, have a surgically inaccessible lesion, or smaller metastasis (<3 cm). For patients with multiple brain metastases or poor performance status, WBRT or supportive care is typically recommended.
Palliative treatment means treatment to shrink a cancer, slow down its growth, or control symptoms caused by the cancer.
Chemotherapy might be used in case of brain metastasis from chemosensitive tumors (eg, small cell lung cancer, lymphoma, choriocarcinoma). However, it has not shown to be beneficial in the management of brain metastasis from NSCLC.
Primary Tumors
The most common intracranial tumors are metastatic rather than primary CNS tumors.
As a general rule, tumors in adults tend to be located above the tentorium while tumors in children tend to be below the tentorium
The three most common primary CNS tumors in adults are:
Glioblastoma
Meningioma
Schwannoma
The three most common primary CNS tumors in children are:
Pilocytic astrocytoma
Medulloblastoma
Ependymoma
In general, when diagnosing primary brain malignancies imaging is first line. If imaging is negative, and suspicion is high, a lumbar puncture should be performed.
Pilocytic astrocytoma
Pilocytic astrocytoma is a type of glioma and is GFAP positive.
Pilocytic astrocytoma most commonly affects young children. Low grade gliomas (including pilocytic astrocytomas) are the most common type of brain tumor in children.
They are most commonly located in the cerebellum but can also appear in the floor and walls of the third ventricle.
Pilocytic astrocytomas characteristically have eosinophilic, corkscrew Rosenthal fibers on histology. Rosenthal fibers can help in the differentiation of pilocytic astrocytomas from other astrocytic gliomas.
These tumors are typically benign and are treated by resection.
Glioblastoma Multiforme
Glioblastoma multiforme is an astrocytoma with WHO grade IV. They have a very poor prognosis with an average of 14 month survival.
Glioblastoma multiforme account for 80% of adult primary brain tumors.
Histopathology of glioblastoma multiforme reveals marked anaplasia (nuclear atypia, pleomorphism) with microvascular proliferation (due to VEGF production) and pseudopalisading necrosis.
Pseudopalisading necrosis is characterized by areas of necrosis and hemorrhage surrounded by rows of malignant cells.
Located within white matter of cerebral hemispheres (often in the centrum semiovale), glioblastoma multiforme can spread to the contralateral hemisphere via the corpus callosum, straddling the cerebral hemispheres to form the classic “butterfly glioma.”
The 4 diagnostic criteria on biopsy are:
Necrosis
Mitosis
Cellular atypia
Neovascularization
Imaging of a patient with glioblastoma multiforme with contrast CT or contrast MRI will reveal butterfly-shaped space-occupying lesion with central necrosis outlined by serpiginous, heterogeneous contrast enhancement.
Treatment of glioblastoma multiforme consists of surgical excision with adjuvant radiation and temozolomide, an alkylating (methylating) agent.
Meningioma
Meningiomas are benign, slow-growing tumors that arise from arachnoid cap cells and are commonly attached to the dura. They can be found in many different locations along the falx cerebri or anywhere on the cerebral convexities.
Meningiomas are benign slow-growing tumors that arise from meningothelial cells (most commonly the arachnoid mater) and are commonly located parasagittally (near the falx cerebri) or near the convexities of cerebral hemispheres.
Meningiomas are the most common type of primary central nervous system tumor.
Meningiomas are most common in older adults (median age at diagnosis of 65 years) and are more common in women.
Risk factors for developing in childhood include neurofibromatosis type 2 and prior radiation exposure (e.g. cranial radiation for leukemia treatment).,
Most meningiomas are asymptomatic and are thus discovered incidentally on neuroimaging. Of the tumors that are symptomatic, seizures are a common manifestation.
Histology shows whorled pattern of spindle cells with psammoma bodies (laminated calcifications).
CT scan and biopsy can make the diagnosis of a meningioma.
Meningiomas are treated with surgery and/or radiation therapy.
Medulloblastoma
Medulloblastoma is the most common malignant intracranial tumor in children and accounts for 20% of all pediatric brain tumors.
Patients with medulloblastoma can present with signs and symptoms of increased intracranial pressure (e.g. morning headache, vomiting, papilledema) and manifestations of cerebellar dysfunction (e.g. nystagmus, ataxia).
Medulloblastoma most frequently arise in the posterior fossa (often in the inferior cerebellar vermis), where they may compress the 4th ventricle and impede CSF outflow via the midline foramen of Magendie and lateral foramina of Luschka leading to hydrocephalus.
Histopathologically, medulloblastomas show Homer-Wright rosettes, which are halos of tumor cells surrounding a central cavity containing neuropil.
Medulloblastomas are treated with a combination of surgery, radiation therapy, and chemotherapy.
Neuroblastoma
Neuroblastoma includes tumors of the sympathetic ganglia and adrenal medulla that are derived from neural crest cells populating these sites.
Neuroblastoma occurs almost exclusively in children and are the most common extracranial solid cancer in infancy.
Neuroblastoma is associated with amplification of the oncogene MYCN (N-myc).
Neuroblastoma can be detected on physical exam of the abdomen; it most commonly presents as a firm,irregular mass that crosses the midline.
In contrast, Wilms tumor most commonly presents as a smooth, unilateral mass.
Neuroblastoma is associated with opsoclonus-myoclonus syndrome, a paraneoplastic syndrome characterized by ataxia, horizontal and vertical saccades (opsoclonus), and rhythmic jerking (myoclonus).
Opsoclonus-myoclonus syndrome is also referred to as “dancing eyes-dancing feet” syndrome.
Ependymomas
In pediatric patients, ependymomas most commonly arise in the posterior fossa, especially within the fourth ventricle. In adults, ependymomas most often occur within the spinal cord.
Patients with posterior fossa ependymomas often present with signs and symptoms of increased intracranial pressure(e.g. morning headache, vomiting, papilledema) due to noncommunicating (obstructive) hydrocephalus.
Recall that ependymomas commonly arise within the fourth ventricle.
Histopathologically, ependymomas show perivascular pseudorosettes with rod-shaped basal bodies (blepharoplasts) near the nucleus.
Pseudorosettes are a spoke wheel arrangement of cells around a lumen. It is thought that these tumor cells are attempting to make "little ventricles".
Craniopharyngiomas
Craniopharyngiomas are benign solid or mixed solid-cystic tumors that arise from remnants of Rathke's pouch.
Craniopharyngiomas are epithelial tumors that usually arise in the pituitary stalk in the suprasellar region, adjacent to the optic chiasm.
Craniopharyngiomas have a bimodal age distribution with a peak in pediatric patients (5-14 years of age) and another peak in adults (>65 years of age).
Clinical manifestations of craniopharyngioma include:
Bitemporal hemianopsia
Headache
Hormonal deficiencies(e.g. growth failure in pediatrics, erectile dysfunction or amenorrhea in adults)
Note: these manifestations are due to compression of the optic chiasm (bitemporal hemianopsia) or the pituitary (hormonal deficiencies).
Diagnosis is suggested via clinical symptoms in association with CT/MRI imaging. The presence of a cystic calcified parasellar lesion is highly suggestive of a craniopharyngioma. Surgery to remove the tumor will confirm diagnosis.
Oligodendroglioma
Oligodendroglioma is a rare, slow-growing tumor that often presents in the frontal lobes of the cerebrum in middle-aged patients.
Histopathologically, oligodendrogliomas show closely packed cells with large nuclei surrounded by clear halo of cytoplasm (fried egg appearance) along with a chicken-wire capillary pattern.
Imaging reveals a calcified tumor in the white matter, usually involving the frontal lobe.
Acoustic Neuroma
An acoustic neuroma, or schwannoma, is an intracranial tumor that arises from the schwann cell sheath of the vestibulocochlear nerve (CN VIII).
Acoustic neuromas represent 80% of all tumors of the cerebellopontine angle, with the remaining 20% being mostly meningiomas.
The presence of bilateral acoustic neuromas is suggestive of neurofibromatosis II.
Symptoms
The most common presentation of an acoustic neuroma is hearing loss in the affected ear(s).
Vertigo and disequilibrium are relatively uncommon presentations, but they can be present.
As the tumor expands, it may compress the facial or trigeminal nerves, causing facial palsy and decreased facial sensation.
Diagnosis
A gadolinium (contrast) enhanced MRI is the diagnostic test of choice for patients with an acoustic neuroma. S100 positive.
Audiometry may still appear on a standardized exam as a diagnostic procedure, but it is no longer used as it has a tendency to miss smaller tumors that have not yet affected hearing.
Treatment
Treatment strategies for managing an acoustic neuroma include:
Observation if the patient is asymptomatic with a small tumor
Stereotactic radiotherapy, which is radiation that is delivered to a precise point or a series of precise points
Surgical excision, which is the treatment of choice for complete tumor removal
Complications associated with acoustic neuromas include cerebellar and brainstem dysfunction due to compression by the tumor.
NF1
Neurofibromatosis type 1, aka von Recklinghausen disease, is an autosomal dominant disease with multiple neurologic tumor and dermatologic manifestations.
Pathogenesis
NF1 is due to mutations in the NF1 gene located at chromosome 17q11.2. Mutations in the NF1 gene result in reduced amounts of the functional protein neurofibromin, causing the wide spectrum of clinical findings, including NF1-associated tumors.
Neurofibromatosis type 1 is the most common type of neurofibromatosis.
Symptoms
The initial signs of neurofibromatosis type 1 include:
Freckling
Cafe-au-lait spots
Lisch nodules
Neurofibromas
Bone abnormalities
Nonunion of bone fragments and fractures during bone development can lead to gait abnormalities, kyphoscoliosis and long bone dysplasia.
Progressive vision loss due to an optic pathway glioma is most commonly seen in children <6 years of age with neurofibromatosis type 1.
Cognitive impairments, seizures, and neuropathic signs are also possible in neurofibromatosis type 1.

Diagnosis
Diagnosis of neurofibromatosis type 1 can be made if they have at least two of the following (remember COFFINS):
>5 Cafe-au-lait macules that are >15mm diameter in postpubertal individuals or >5mm in prepubertal individuals
Optic glioma
Axillary or inguinal Freckling
first degree relative with neurofibromatosis type 1 (Family history)
≥2 Iris hamartomas (Lisch nodules)
≥2 Neurofibromas or 1 plexiform neurofibroma
Bone/ Skeletal lesions
MRI will show multiple areas of increased signal intensity in the brain and increased brain volume.
Treatment
Treatment of neurofibromatosis type 1 is aimed at maintaining function and treating complications.
Complications of neurofibromatosis include:
Risk of malignant CNS tumors
Developmental delays
Mental retardation
Peripheral neuropathy
Pheochromocytoma
Vision abnormalities
Bone abnormalities
NF2
Pathogenesis
Neurofibromatosis type 2 is a dominantly inherited syndrome that predisposes patients to multiple tumors of the central nervous system.
Neurofibromatosis type 2 is less common than neurofibromatosis type 1.
Neurofibromatosis type 2 (NF2) is consistently linked with abnormalities of the NF2 gene, which is located on chromosome 22 and produces the protein merlin.
Development of schwannomas in individuals with neurofibromatosis type 2 requires inactivation of both NF2 alleles.
Symptoms
Patients with neurofibromatosis type 2 present around the age of 20 with:
Bilateral acoustic neuromas
Multiple meningiomas
Neurofibromas (less common than type I)
Cataracts
The diagnosis of neurofibromatosis type 2 consists of possessing one of the following criteria:
Bilateral vestibular schwannomas
First degree relative with neurofibromatosis type 2 AND unilateral vestibular schwannoma or any two of the following:
Meningioma
Schwannoma
Glioma
Neurofibroma
Posterior subcapsular lenticular opacities
Multiple meningiomas with unilateral schwannomas or any two of the following:
Glioma
Neurofibroma
Schwannoma
Cataract
Treatment
The goal of treatment in patients with neurofibromatosis revolves around maintaining quality of life and preservation of function.
Stereotactic radiosurgery (high single dose of radiation) and stereotactic radiotherapy (focused doses of radiation over several treatment sessions) have become important modalities in the treatment of appropriately selected patients with sporadic vestibular schwannomas.
Sturge-Weber
Pathogenesis
Sturge-Weber Syndrome (encephalotrigeminal angiomatosis) is a rare congenital vascular disorder characterized by facial capillary malformation (port wine stain) and associated capillary-venous malformations affecting the brain and eye.
Sturge weber is caused by somatic mosaic mutations and thus is not a heritable disorder.
Symptoms
The most common presentation of Sturge-Weber syndrome is the port-wine stain nevus flammeus “birthmark”. This is a macular vascular lesion (congenital unilateral capillary or cavernous hemangioma) on the face in the dermatomal distribution of cranial nerve V — usually the V1 or V2 dermatomal distribution. It does not blanch with pressure.
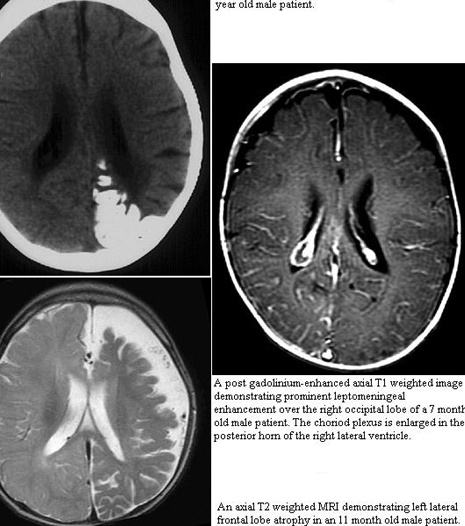
Some patients have an ipsilateral arteriovenous malformation of pia mater vessels (ipsilateral leptomeningeal angioma) overlying occipital and parietal lobes.
Children often suffer from visual disturbances (eg, hemianopia) and often present with buphthalmos (markedly enlarged eye) due to congenital glaucoma that leads to retained aqueous humor.
Children develop progressive neurologic deterioration:
Seizures
Mental retardation
Hemiparesis
Hemisensory deficit
Diagnosis
The diagnosis of Sturge-Weber syndrome is based upon demonstration of the facial capillary malformations and leptomeningeal capillary-venous malformations.
MRI with contrast will reveal the presence of the leptomeningeal capillary-venous malformation and the extent of involvement with brain structures.
Treatment
There is no specific treatment for Sturge-Weber syndrome and management revolves around controlling seizures and glaucoma as they present in the patient.
Tuberous Sclerosis
Pathogenesis
Tuberous sclerosis is an inherited neurocutaneous disorder that presents with benign hamartomas of multiple organ systems.
Tuberous sclerosis is an autosomal dominant disorder caused by mutation in tumor suppressor genes TSC1 or TSC2.TSC1 codes for the protein hamartin and TSC2 codes for the protein tuberin.
Symptoms
Tuberous sclerosis most often presents in infancy with:
Infantile spasms (epilepsy)
Autism
Neonatal cardiac rhabdomyomas
Classic exam findings of tuberous sclerosis can be remembered with AASS RRRASH:
Ash leaf spots (hypopigmented spots found on the skin)
Angiofibromas (reddish brown papules)
Shagreen patches (rough patches with orange peel consistency)
Seizures
Mental retardation
Retinal lesions, such as mulberry tumors or phakomas (round, flat, gray lesions located on the periphery of the retina)
Rhabdomyomas
Angiomyolipomas of the kidney
Subependymal astrocyte proliferation in the brain (sometimes referred to as "candle guttering")
Hamartomas
Diagnosis
Tuberous sclerosis is a clinical diagnosis based on the exam findings above. While not necessary to make the diagnosis, genetic testing can confirm the genetic mutation of TSC1 or TSC2.
Management of tuberous sclerosis revolves around controlling symptoms, such as seizure control, and periodic imaging to monitor for malignancies.
Last updated