Brain
Cerebral Palsy
Cerebral palsy is a heterogeneous set of disorders characterized by motor deficits and postural dysfunction caused by cerebral motor deficits that are acquired during development before the age of 2 or 3 (depending on source).
Can be early prenatal, perinatal, or postnatal injury due to vascular insufficiency; toxins or infections; or the pathophysiologic risks of prematurity.
Types of motor deficits:
Spastic type results in stiff muscles and hyperreflexia
Dyskinetic type is a dystonic or ataxic movement disorder
Prevalence of cerebral palsy is 2:1000 births with increased prevalence among babies with low birth weight (5-15% of surviving infants with very low birth weights suffer from cerebral palsy).
Risks
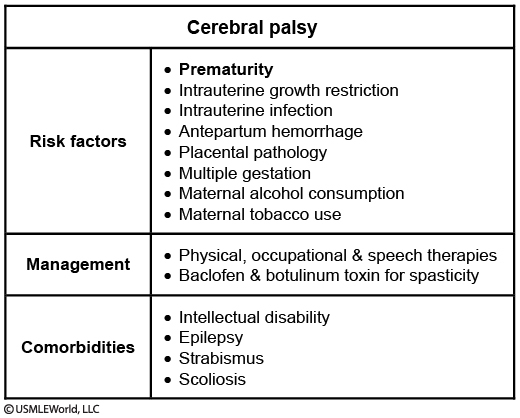
Risk factors for having a baby with cerebral palsy:
Intrauterine growth restriction
Prematurity
Severe placental pathology
Multiparous pregnancies
Perinatal hypoxia
Intrauterine infection
Symptoms
Cerebral palsy presents differently depending on which type the patient has. However, despite each type having their own associated symptoms, some patients may present with symptoms of both spastic and dyskinetic motor deficit.
Patients with spastic type will present with:
Increased tone
Increased deep tendon reflexes
Gait abnormalities
Mental Retardation
Patients with dyskinetic type present with choreoathetoid and ataxic movementsthat worsen with stress.These patients also havetrouble speaking.
Both types of cerebral palsy can present with seizures.
Diagnosis
Cerebral palsy is a diagnosis of exclusion. One must exclude all neurodegenerative disorders, inborn metabolic errors, neoplasms, seizures/epilepsy, and neuromuscular disorders.
MRI may be useful in determining causative lesions of cerebral palsy.
Treatment
Treatment and prognosis vary greatly from patient to patient and treatment plans are tailored to each patient's condition. Pharmacologic therapy, such as botulinum toxin, dantrolene, baclofen and benzodiazepines, can decrease spasticity in the patient. Physical therapy can be used to alleviate symptoms and improve function.
Friedreich's ataxia
Pathogenesis
Friedreich’s ataxia is a hereditary ataxia resulting in motor incoordination due to cerebellar dysfunction.
The mutation responsible for Friedreich’s ataxia is an autosomal recessive loss of function mutation in the frataxin (FXN) gene on chromosome 9, usually the result of a GAA trinucleotide repeat.
Frataxin is a mitochondrial protein responsible for many interactions with iron. Levels of this protein are elevated in Friedreich’s ataxia due to dysfunction of the protein. Dysfunction of this protein is thought to cause oxidative stress leading to the symptoms of Friedreich’s ataxia.
The severity of manifestations correlates with an increasing number of trinucleotide repeats.
Symptoms
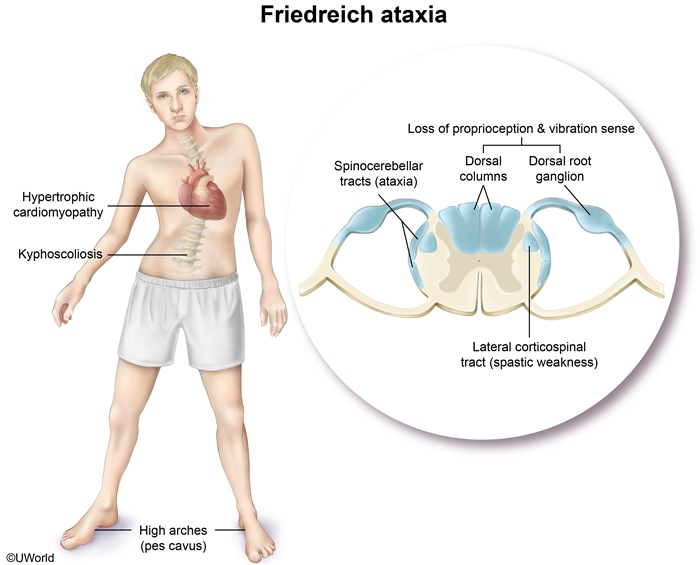
Hypertrophic cardiomyopathy is the common cardiac finding in patients with Friedreich’s ataxia, resulting in arrhythmia, and heart failure.
Spinocerebellar and lateral corticospinal tract degeneration causes gait ataxia and spastic muscle weakness, respectively.
Degeneration of the dorsal columns and dorsal root ganglia causes loss of position and vibration sensation. Also loss of reflex.
Kyphoscoliosis and foot abnormalities (pes cavus) are characteristic skeletal deformities. Atrophy of small muscles of hands.
Heart involvement includes hypertrophic cardiomyopathy and congestive heart failure.
Diabetes mellitus develops in about 10% of patients with Friedreich ataxia.
Diagnosis
Friedreich’s ataxia is diagnosed based on clinical findings and genetic testing.
Management
Patients with Friedreich’s ataxia can be treated with beta-blockers in order to reduce the symptoms associated with hypertrophic cardiomyopathy, and antioxidants (such as coenzyme Q10) to decrease markers of oxidative injury. However, no treatment has been found to reduce the progression of the neurological symptoms.
The most common cause of death for patients with Friedreich’s ataxia is congestive heart failure or dysrhythmia.
Multiple Sclerosis
Multiple Sclerosis (MS) is a chronic neurologic condition of white matter lesions separated by time (acute and past symptoms) and space (different parts of the CNS affected), marked by unpredictable relapses with long asymptomatic remissions.
It begins most commonly in young adulthood, affecting women twice as often as men with a peak incidence between 20 and 30 years of age.
Pathogenesis
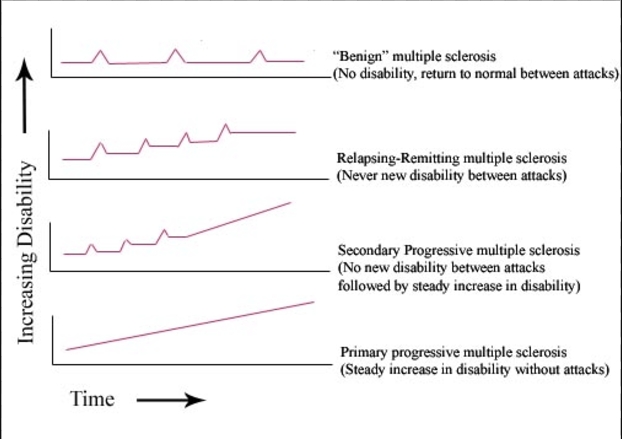
There are 4 subtypes of multiple sclerosis:
Relapsing-Remitting (most common subtype): clearly defined relapses of disease symptoms followed by complete recovery.
Clinically isolated symptoms: first attack of a disease compatible with MS but does not fulfill diagnostic criteria.
Secondary progressive: initially relapsing-remitting disease followed by gradual worsening symptoms with or without occasional relapses, minor remissions, and plateaus.
Primary progressive: characterized by progressive accumulation of disability from disease onset.
While the cause is unknown, multiple sclerosis is characterized by autoimmune demyelination of oligodendrocytes in the CNS, histologically characterized by sharply defined area of myelin loss with relative preservation of axons. Peripheral nerves are NOT affected, only the CNS (note: the optic nerve is an extension of the CNS, not a true peripheral nerve).
The risk of developing MS is associated with certain alleles of the major histocompatibility complex (MHC), particularly the HLA-DRB1 locus.
Multiple sclerosis is more common in northern latitudes. Those who move from a low-risk to a high-risk geographic region before the age of 15 adopt the risk associated with their new home, while those who migrate after 15 retain risk of childhood home.
Pregnancy is associated with decreased relapse frequency, especially during the 3rd trimester.
Symptoms
Multiple sclerosis typically presents in a young adult with a clinically isolated syndrome suggestive of MS such as optic neuritis, spinal cord (eg.sensory sensation and motor function) and brainstem syndrome (eg, internuclear ophthalmoplegia).
Optic neuritis:
usually first sign and can present with blurry vision or sudden painful loss of vision. Physical examination of patients with optic neuritis can reveal a relative afferent pupillary defect (Marcus-Gunn pupil).
Spinal Cord:
Sensory dysfunction (paresthesias, loss of pain and temperature sensation, loss of vibratory sensation)
Upper motor neuron dysfunction
Autonomic dysfunction (urge incontinence due to hyperactive detrusor muscle),
Lhermitte phenomenon: flexion of the neck produces electrical sensation down spine.
Brainstem:
Scanning speech (sound drunk)
Intention tremor
Cerebellar ataxia
Cerebellar ataxiaInternuclear ophthalmoplegia (bilateral demyelination of MLF)
Nystagmus
Symptoms may worsen with heat (e.g. bath, warm weather) because heat slows rate of conduction of electrical activity through demyelinated nerves even more. This is called Uhthoff's phenomenon.
Multiple sclerosis (MS), an autoimmune demyelinating central nervous system disorder, is one of the few conditions that may present with trigeminal neuralgia bilaterally. This occurs due to demyelination of the nucleus of the trigeminal nerve or the nerve root, which leads to improper signaling of the nerve and paroxysms of severe pain. This patient's episode of right hand numbness that lasted 2 weeks and spontaneously improved was likely her first symptom of MS.
Diagnosis
Multiple sclerosis is a clinically based diagnosis supported by imaging and CSF analysis.
MRI is the gold standard for diagnosis because of increased sensitivity in detecting demyelinating plaques, revealing periventricular plaques (areas of oligodendrocyte loss and reactive gliosis) with preservation of axons.
The detection of oligoclonal IgG bands with isoelectric focusing is the most important diagnostic CSF study for multiple sclerosis (MS).
An elevated CSF IgG index is also supportive of MS.
Treatment
Treatment for MS falls into three categories: acute therapies for relapses (e.g. corticosteroids), chronic therapies that treat the underlying disease process (e.g. β Interferon), and symptomatic therapies (e.g. baclofen, an anti-spasmodic).
Acute attacks of multiple sclerosis are treated with high dose IV corticosteroids ( methylprednisolone ). This treatment will shorten the duration of attack but doesn’t alter progression of disease. Without any treatment, symptoms of the acute attack resolve in 6 weeks. Therapeutic plasma exchange can be used in steroid resistant cases.
Disease-modifying therapy for multiple sclerosis includes:
Beta-Interferon therapy increases integrity of blood-brain-barrier reducing the relapse rate by 30%-40%. Check CBC and liver function tests since interferon therapy can lead to leukopenia and transaminitis.
Natalizumab is a monoclonal antibody that binds α4-subunit of integrins on WBCs disrupting interaction with VCAM-1 (vascular cell adhesion molecule-1) and MAdCAM-1 (mucosal addressin cell adhesion molecule-1) ultimately preventing WBCs from exiting the blood vessels to subjacent inflamed tissues
Glatiramer has an immunomodulatory mechanism of action that involves binding to major histocompatibility complex molecules and consequent competition with various myelin antigens for their presentation to T cells.
For rapidly progressive disease and disease refractory to other treatments: Cyclophosphamide, methotrexate, azathioprine.
Symptomatic control of multiple sclerosis is accomplished with:
Muscle spasticity using baclofen, diazepam, tizanidine
Neuropathic pain using carbamazepine or gabapentin
Bladder dysfunction using anticholinergics
Neuromyelitis optica
Neuromyelitis optica (also called Devic disease) is an inflammatory disorder similar in presentation to multiple sclerosis that is characterized by immune-mediated demyelination of the optic nerves (resulting in vision loss and eye pain exacerbated by eye movement) and spinal cord (resulting in limb weakness, bladder dysfunction, and sensory impairment).
Neuromyelitis optica can be distinguished from multiple sclerosis by the presence of aquaporin-4 (AQP4) autoantibodies (also known as NMO-IgG) in the serum and CSF patients with neuromyelitis optica.
Normal Pressure Hydrocephalus
Normal pressure hydrocephalus is a collection of excess cerebrospinal fluid in the cerebral ventricles and spinal cord without an increase in intracranial pressure.
Causes of normal pressure hydrocephalus can include subarachnoid hemorrhage, chronic meningitis, or any other cause of impaired CSF resorption.
Symptoms
To remember the symptoms of normal pressure hydrocephalus, use the mnemonic wacky (cognitive impairment), wobbly (gait abnormalities), and wet (incontinence).
Diagnosis and Management
A levodopa challenge can be administered in normal pressure hydrocephalus in order to rule out idiopathic parkinsonism as patients with NPH have no significant response.
Normal pressure hydrocephalus shows normal pressure on lumbar puncture despite improvement of symptoms with cerebrospinal fluid removal.
MRI of a patient with normal pressure hydrocephalus shows widened cerebral ventricles. White matter lesions and cerebral aqueduct atrophy can also be seen.
The treatment of normal pressure hydrocephalus involves ventriculoperitoneal shunting.
Dyslexia
Dyslexia is a learning disability secondary to neurological disorders in children with normal vision and intelligence that manifests as a reading difficulty.
Research has shown a genetic component to dyslexia, as it tends to run in families. Other risk factors for dyslexia include:
A family history of reading speech problems
History of speech and language impairment in preschool.
Learning new words slowly
Late talking
Symptoms
It is characterized by difficulties with accurate and/or fluent word recognition and by poor spelling and decoding abilities.
Dyslexia will present as a child with normal to high IQ who starts having difficulty with reading assignments in kindergarten/elementary school. As school progresses, children with reading disability may present with a decline in school performance or difficulty keeping up.
Children who have a reading disability may have associated deficits in attention, executive function, and social skills that can further interfere with performance in school.
Diagnosis and management
Dyslexia is a clinical diagnosis. It is important to exclude any vision, hearing or cognitive impairments prior to making the diagnosis.
Management of reading disability may involve educators, psychologists, and pediatric clinicians. Interventions are usually provided by educators within the school system and by private tutors.
BPPV
Benign paroxysmal positional vertigo is the most common cause of vertigo.
Benign paroxysmal positional vertigo is most commonly caused by the movement of otoconia (calcium carbonate crystals) within the semicircular canals due to changes in head position. The presence of otoconia within the semicircular canal is known as canalithiasis.
The movement of the otoconia within the semicircular canal causes transient vertigo and nystagmus.
Benign paroxysmal positional vertigo is most common in women over age 50.
Symptoms
Benign paroxysmal positional vertigo is elicited by changes in head position relative to gravity, such as:
Lying down
Turning over in bed
Tilting the head to look upward
Patients with benign paroxysmal positional vertigo typically present with the following symptoms:
Vertigo (dizziness and spinning) that is paroxysmal nature (short lived) and inducible by changing position
Nausea
Horizontal nystagmus
In contrast to Ménière disease, hearing loss and tinnitusare absent in patients with benign paroxysmal positional vertigo.
Diagnosis
A Dix-Hallpike maneuver will be positive during physical exam of patients with benign paroxysmal positional vertigo, which is where turning the patient’s head side to side while quickly moving from sitting to supine induces both vertigo and horizontal nystagmus.
If clinical findings are not sufficient for the diagnosis of paroxysmal positional vertigo, normal TSH levels can rule out thyroid pathology and a CT or MRI helps rule out intracranial lesions.
Treatment
Treatment of benign paroxysmal positional vertigo is aimed at reducing recurrent episodes and involves the use of physical maneuvers to free otolith crystals from the posterior semicircular canal. These treatments are called Epley and Semont maneuvers. Home or self exercises are called Brandt-Daroff exercises but are generally less effective than Epley and Semont maneuvers.
Meniere Disease
Meniere disease is a condition of the inner ear that causes distension of the endolymphatic sac (endolymphatic hydrops).
Distension of the endolymphatic sac in Meniere disease is thought to occur due to either a blockage of the endolymphatic channel or increased endolymphatic fluid production.
Meniere disease has a median age of onset of 50 years of age.
Symptoms
It results in episodic bursts of vertigo and tinnitus.
Symptoms of Meniere disease include:
Episodic vertigo
Sensorineural hearing loss: worsening overtime
Tinnitus
Sensation of ear fullness/pressure
Nausea and vomiting
Physical exam findings in Meniere’s disease include a negative Dix-Hallpike positional test (which is positive in benign paroxysmal positional vertigo) and possible production of symptoms with pneumo-otoscopy.
Diagnosis
Audiometry shows low-frequency hearing loss early in the course of Meniere disease.
When working-up Meniere disease, make sure to rule out:
Perilymphatic fistula (abnormal communication between the perilymphatic space and the middle ear or mastoid)
Multiple Sclerosis
Acoustic Neuroma
Hyper- and hypothyroidism
Treatment
Patients with Meniere disease should be encouraged to limit intake of salt, caffeine, nicotine, and alcohol.
Agents used to treat acute episodes of Meniere disease include:
Anticholinergics
Antiemetics
Antihistamines
Benzodiazepines
Patients with Meniere disease that do not respond to dietary modifications can be treated with diuretics.
Intratympanic gentamicin and surgery can be used for patients with severe Meniere disease that is refractory to medical treatment.
Recall that aminoglycosides are toxic to vestibular hair cells.
Complications
The most common complication of Meniere’s disease is a progressive hearing loss at all frequencies.
Last updated